
Amide-imide tautomerism of acetohydroxamic acid in aqueous solution: quantum calculation and SMD simulations†
S. Tolosa*a,
N. Mora-Diez*b,
A. Hidalgoa and
J. A. Sansóna
aDepartamento de Ingeniería Química y Química Física, Universidad de Extremadura, Badajoz, Spain. E-mail: santi@unex.es
bDepartment of Chemistry, Thompson Rivers University, Kamloops, BC V2C 0C8, Canada. E-mail: nmora@tru.ca
First published on 3rd September 2014
Abstract
The kinetics and the thermodynamics of the amide-imide tautomerizations of acetohydroxamic acid in aqueous solution are studied from a theoretical point of view. Quantum electronic structure calculations (QCISD//MP2, MP2 and M06-2X, applying two continuum solvation methods) and steered molecular dynamic (SMD) simulations (with solute–solvent interaction potentials derived from AMBER van der Waals parameters and electrostatic charges in solution) are taken into account. The elementary and complex tautomerizations from the E-Amide and Z-Amide forms are studied with and without the explicit assistance of a water molecule. The Gibbs free energy of activation for the E-Amide ⇆ E-Imide and Z-Amide ⇆ Z-Imide processes are considerably reduced when these processes are assisted by one water molecule. The Gibbs free energy of reaction was also reduced, but to a lesser degree, for each of the processes considered when in the presence of an explicit solvent molecule. The Z-Amide ⇆ Z-Imide tautomerization, which occurs in two elementary steps, seems to be slightly more favoured from a thermodynamic point of view; however, the E-Amide ⇆ E-Imide tautomerization, which is an elementary process, is the most kinetically favoured. The SMD simulations led to results which are consistent with those obtained by the three electronic structure methods applied.
1. Introduction
Theoretical studies of chemical and biochemical reactivity in solution are of research interest.1–9 The amide-imide tautomerization of hydroxamic acids in aqueous solution has been previously studied,10–33 often using formohydroxamic and acetohydroxamic acids as models to understand this process of great importance in chemistry and biochemistry which remains of interest. Hydroxamic acids have the additional complexity of also being involved in E-Z isomerization. Hence, ignoring the deprotonation processes that take place in solution, for each hydroxamic acid there are four species to consider (see Fig. 1). The significantly less thermodynamically stable nitroso form (an α-nitroso alcohol isomer)10 has been excluded from this study. | ||
| Fig. 1 Most-stable conformations of the acetohydroxamic acid isomers in aqueous solution calculated at the QCISD-PCM/cc-pVDZ and MP2-PCM/6-311++G(d,p) levels of theory at 298.15 K (relative Gibbs free energies of formation are shown in parentheses: QCISD, MP2). | ||
How are the isomers of hydroxamic acids in aqueous solution related from both thermodynamic and kinetic points of view? A good description of the isomerization processes requires an effective modeling of these systems in solution. Quantum electronic structure calculations making use of continuum solvent models, with or without explicit solvent molecules, are a frequently used methodology to study systems in solution. However, molecular dynamic (MD) simulations are a desirable alternative approach for predicting the evolution of these processes in time and to account for the molecular description of the solvent.
One of the main difficulties of theoretical studies in solution comes from the great number of atoms involved and the number of molecular interactions with the surrounding environment that must be accounted for. In simulating biochemical and chemical processes in dilute aqueous solutions, our research group uses the classical method of molecular dynamics. In previous studies,34–40 we have described the solute–solvent interaction through LJ (12-6-1) analytical functions, whose molecular parameters are obtained by fitting ab initio interaction energies or by using AMBER force fields41 for the solute, and the TIP3P potential42 for the water–water interactions. Having described the potentials, one proceeds to simulate the system by applying the statistical methods of molecular dynamics. The acceleration of the process is achieved by using the steered molecular dynamic (SMD) technique,43,44 which applies external steering forces in the right direction (which in the present study are mainly on the hydrogen transferred in the process that moves throughout a reaction coordinate). Although this technique has been applied to the study of conformational or configurational changes of biological systems in the gas phase,45–48 it can also be used for solvent-explicit calculations, as in the case of intramolecular proton transfers in water.
Although there are numerous theoretical studies related to hydroxamic acids in solution, most of them deal with the deprotonation of formohydroxamic and acetohydroxamic acids in the gas phase using electronic structure calculations. The amide-imide tautomerization in solution has been much less studied, and MD simulations have not been previously performed to study this process. Tavakol10 performed the computational study of the water-assisted mechanism for the Z-Amide ⇆ Z-Imide process for several hydroxamic acids at the B3LYP/6-311++G(d,p) level of theory (however, the transition state reported corresponds to the E-Amide ⇆ E-Imide process). It was found that the explicit presence of one water molecule (without the use of a continuum solvation method) barely changes the gas-phase reaction energy, but significantly reduces the energy of activation of this process. The addition of a second water molecule was only able to decrease the Gibbs free energy of activation by 0.5 kcal mol−1; the presence of a third water molecule increased the value relative to the result obtained with one and two explicit solvent molecules. Senent et al.11 and Mora-Diez et al.12 using MP2/AUG-cc-pVDZ calculations found that although in the gas phase the N-deprotonation of acetohydroxamic acid in its Z-Amide form is the most favourable, the energy associated with the N-deprotonation of the E-Amide tautomer in solution is very similar, so the Amide ⇆ Imide tautomerism between the E isomers should also be considered. Saldyka and Mielke13 studied the tautomerism between different E- and Z-isomers of acetohydroxamic acid in the gas phase at the MP2/6-311++G(d,p) level of theory from a thermodynamic point of view, finding the Z-structure to be the most stable. Fitzpatrick and Mageswaran14 addressed the stability, structure and atomic populations of the four isomers of acetohydroxamic acid with ab initio calculations, finding that the E-Amide form is more stable than the Z-Amide form, followed by the Z-Imide and E-Imide isomers, while for the systems with a water molecule, Z-Amide is the most stable isomer. Kakkar et al.15 studied the conformational behaviour of some hydroxamic acids via different mechanisms at the B3LYP/6-311++G(d,p)//B3LYP/6-31G(d) level of theory, obtaining results showing that in solution the amide forms are more stable and that the Z-Amide isomer is only slightly more stable than the E-Amide. Other studies on the isomers of hydroxamic acids have been performed in gas phase, with results varying depending on the level of calculation.16–33
The tautomeric equilibria of acetohydroxamic acid (AHA), of special importance in biochemistry, are very sensitive to the medium where the reactions occur. Therefore, AHA is an appropriate system for the study of the amide-imide tautomerization process via different mechanisms. The Amide ⇆ Imide tautomerization reaction takes place as a result of the intramolecular proton transfer between the amide and imide forms, which initially requires an adequate orientation of the hydrogen atom bonded to nitrogen, since this site is the most favourable for amide deprotonation.12 Even though the Z-Amide form of acetohydroxamic acid is the most stable one in aqueous solution from a thermodynamic point of view, the E-Amide form is only 0.05 (1.16) kcal mol−1 higher in Gibbs free energy when calculated at the QCISD-PCM/cc-pVDZ (MP2-PCM/6-311++G(d,p)) level of theory (see Fig. 1). Hence, the E-Amide ⇆ E-Imide transformation is also an important process to be considered in solution. The amide-imide transformations in acetohydroxamic acid should be expected to occur in single or multiple kinetic steps depending on the structural similarity of the isomers considered.
This article focuses on the thermodynamic and kinetic study of the elementary and complex tautomerizations of the E-Amide and Z-Amide forms of acetohydroxamic acid in aqueous solution, with and without the presence of an explicit solvent molecule. The calculations reported in this article, applying two quantum electronic structure methods and using mean force potentials from steered molecular dynamic (SMD) simulations, are analyzed and compared for several mechanisms in aqueous solution. Given that to the best of our knowledge, MD simulations have not been previously applied to study the amide-imide tautomerism, and that a complete thermodynamic and kinetic study of the tautomeric equilibria in AHA has not been previously reported, the results reported in this paper are additionally relevant.
2. Computational details
2.1. Electronic structure calculations
The most stable conformations of the different species considered in this study have been calculated using the GAUSSIAN 09 program.49 Geometries have been fully optimized and characterized at the MP2/6-311++G(d,p) level of theory. Solvent effects are accounted for on the geometry optimizations and frequency calculations by means of the IEF-PCM method,50 employing the UFF atomic radii when constructing the solvent cavity. Additional calculations have been performed using the M06-2X51 functional and the 6-311++G(d,p) basis set in combination with the SMD52 solvation method on both geometry optimizations and frequency calculations. Calculations at the QCISD-PCM/cc-pVDZ//MP2-PCM/6-311++G(d,p) level of theory were also performed.2.2. MD simulations
To describe the solute–solvent interaction potential, a LJ (12-6-1) function is used (see eqn (1)).
 | (1) |
The potential of mean force (PMF) in a reaction represents the Gibbs free energy change with the reaction coordinate. To relate Gibbs free energy differences between two equilibrium and non-equilibrium processes, we use the Jarzynski's equality.54 Thus, this Gibbs free energy change, ΔG, is related to an average of all possible works 〈W〉 of an external process that take the system from the equilibrium state A to a new state B, as shown by eqn (2):
 | (2) |
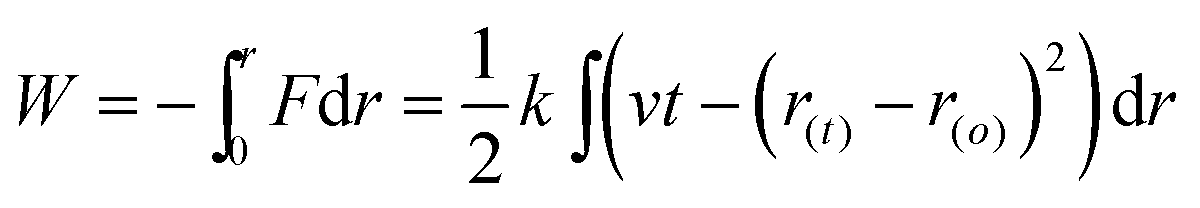 | (3) |
The molecular dynamics simulations of an NVT ensemble with periodic boundary conditions for each acetohydroxamic structure in an aqueous environment, formed by about two hundred molecules depending on the system considered, were carried out at 298.15 K using the AMBER program.55 The time considered for the different simulations was 2 ns with time steps of 0.5 fs. The first 1000 ps were taken to ensure that equilibrium was completely reached. This can be confirmed by considering that the standard deviation of the potential energy values in the simulation was lower than 1% of the mean values in all the cases. The last 1000 ps were stored every 100 fs to obtain the trajectory of the simulated molecule. The water molecules initially located at distances less than 1.6 Å from any solute atom were eliminated from the simulations. The long-range electrostatic interactions were treated by the Ewald method.56 A cut-off of 7 Å was applied to the water–water interactions to simplify the calculations, and periodic boundary conditions were used to maintain the number of solvent molecules constant.
The steered molecular dynamics (SMD) simulations allow the calculation of standard Gibbs free energies of activation (ΔG≠) and reaction (ΔG) from potential of mean force (PMF), since they follow the time-evolution of the process, i.e., going from a reactant configuration, to the TS, and, finally, to the product of a particular elementary step, focusing on one or more reaction coordinates. Approximate structures of the stationary points involved in a reaction mechanism can be obtained.
In order to perform the SMD simulations, the QM/MM method with the semi-empirical QM Hamiltonian SCC-DFTB,57,58 implemented in the AMBER12 software,59,60 was used. The system was initially minimized with a 1 ps simulation. Subsequently, the system was equilibrated at 200, 400, 600, 800, and 1000 ps (with 0.5 fs time steps) in order to obtain different paths for each SMD simulation. Finally, during the last 1000 ps of the simulation, all the atoms of acetohydroxamic acid were allowed to move freely. The force constant used was 1000 kcal mol−1 Å−2 for the distance and 10 kcal mol−1 rad−2 for the torsion, and the amidic hydrogen atom was directed from the amide to the imide structure. The reaction coordinates chosen for the non-assisted and water-assisted mechanisms are discussed in Section 3.1.1.
2.3. Thermodynamic and kinetic calculations
In addition to reporting standard enthalpies (ΔH°) and Gibbs free energies (ΔG°) of reaction for each of the processes considered, equilibrium constants (K) are calculated using eqn (4).
 | (4) |
Rate constants (k) for elementary steps are calculated applying classical transition state theory (TST) using eqn (5), where κ is the tunneling factor, kB is Boltzmann's constant, h is Planck's constant, R is the ideal gas constant, and ΔG≠ is the standard Gibbs free energy of activation for a particular elementary step, which is calculated by subtracting the standard Gibbs free energies of formation of the transition state and the reactant. The rate constant for the corresponding reverse reaction can be calculated using eqn (6).
 | (5) |
 | (6) |
The tunneling factor is calculated by assuming an asymmetrical Eckart barrier61 for each of the processes in which a hydrogen ion is transferred. This calculation uses the enthalpy of activation (ΔH≠) and the enthalpy change of the forward reaction at 298.15 K, as well as the imaginary frequency of the TS. A modified version of the numerical integration program of Brown62 is used for the calculation of the tunneling factor.
3. Results and discussion
3.1. Amide-imide tautomerizations: mechanisms considered
The E-Amide ⇆ E-Imide and the Z-Amide ⇆ Z-Imide processes were studied with and without the presence of an explicit solvent molecule, in addition to accounting for additional solvent effects when applying a continuum method or during the MD simulations. While the two E-Amide ⇆ E-Imide transformations are elementary, the two Z-Amide ⇆ Z-Imide processes, as well as the Z-Amide ⇆ E-Imide transformation, are complex (two elementary kinetic steps are required). The five mechanisms considered and the acronyms used to identify the species involved, including the transition states, are shown in Fig. 2. While a brief description of these processes follows, some structural details of the species involved are discussed in Section 3.2.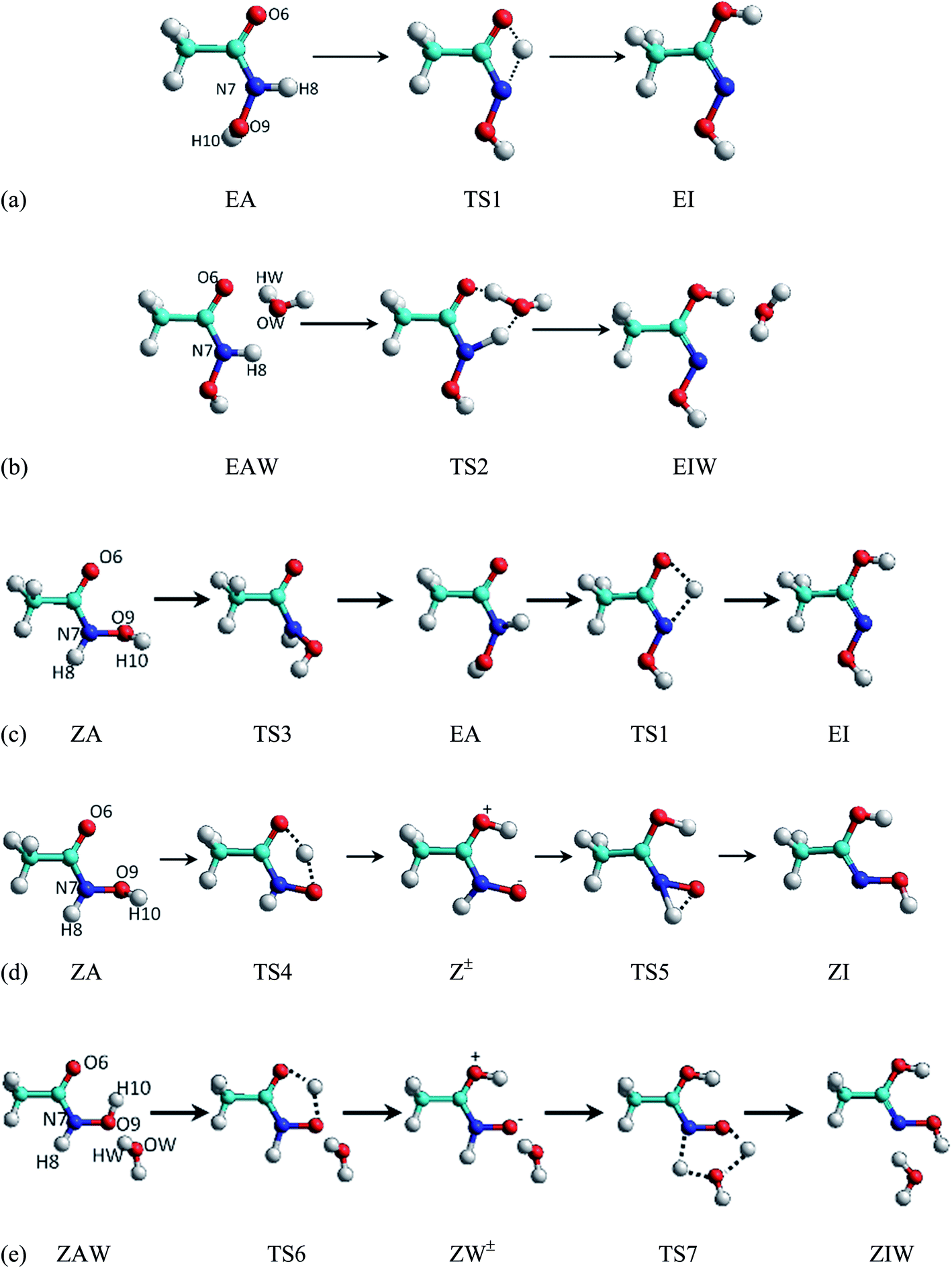 | ||
| Fig. 2 Elementary and complex amide ⇆ imide tautomerizations studied (the labels shown on the reactant species are used to identify the reaction coordinates followed during the SMD simulations: see Section 3.1.1.). | ||
In the non-assisted mechanism, the amidic hydrogen of the E-Amide (EA) isomer is transferred to the carbonyl oxygen via a four-member-ring transition state (TS1) before forming the E-Imide (EI) isomer (see Fig. 2a). In the water-assisted mechanism (see Fig. 2b), the amidic hydrogen in the E-Amide-water complex (EAW) is transferred to the water oxygen and, simultaneously, a water hydrogen is transferred to the carbonyl oxygen via a six-member-ring transition state (TS2) prior to forming the E-Imide-water complex (EIW). In the non-assisted mechanism from Z-Amide (ZA) to E-Imide (EI) (see Fig. 2c), there is first an internal rotation around the C–N single bond going through TS3 to form EA. Following this step, TS1 connects EA with its tautomer EI, as previously described. Because the barrier to internal rotation from the Z-Amide complex (ZAW) to EAW would be much greater than for ZA ⇆ EA because of the presence of the explicit solvent molecule, this transformation was not studied.
The unassisted conversion from Z-Amide (ZA) to Z-Imide (ZI) (see Fig. 2d) involves two steps. First, the hydroxide hydrogen is transferred to the carbonyl oxygen via a five-member-ring transition state (TS4) to form a zwitterion intermediate Z±. From this point, the second proton transfer, from the nitrogen to the oxygen close to it, takes place via TS5 to form ZI. The water-assisted transformation of the Z-Amide complex (ZAW) to form the Z-Imide complex (ZIW) (see Fig. 2e) initially goes through a transition state (TS6) in which the hydroxide hydrogen is transferred to the carbonyl oxygen to form the intermediate zwitterion water complex ZW±. In the following kinetic step, a concerted double proton-transfer, from the amidic nitrogen to the water oxygen and from this atom to the previously deprotonated oxygen, takes place via a six-member-ring transition state (TS7) to produce ZIW.
The process followed when studying the complex mechanisms (c), (d) and (e) was to initially focus on the first step in the forward direction (i.e., ZA → EA, ZA → Z± and ZAW → ZW±), then focus on the second step in the reverse direction (i.e., EI → EA, ZI → Z± y ZIW → ZW±), and combine them.
3.2. Comments on structural aspects
Fig. 3 shows the atomic charges (in bold on atoms) and some important geometric parameters of the MP2-PCM/6-311++G(d,p) most stable conformations of the reactants, intermediates and products involved in the tautomerizations studied. Similar information for the calculated transition states (TSs) is displayed in Fig. 4. The structures of the TSs were verified by carrying out a normal mode analysis, which provides a single imaginary frequency (see values at the bottom of each structure) and whose eigenvector corresponds to the transformations taking place in each of the processes previously described. Further confirmation of the correctness of the calculated TSs was obtained after performing IRC calculations which properly connected each TS with the corresponding reactant and product species. The Cartesian coordinates for each of these stationary points are reported in the ESI† section. | ||
| Fig. 3 Important geometric parameters and atomic charges of the reactants, intermediates and products calculated at the MP2-PCM/6-311++G(d,p) level of theory. | ||
 | ||
| Fig. 4 Important geometric parameters and atomic charges of the calculated transition states calculated at the MP2-PCM/6-311++G(d,p) level of theory. | ||
In going from EA to TS1, the C–O and N–H distances increase by 0.07 and 0.30 Å, respectively. The final O–H distance in EI is 0.97 Å. In going from EA to EI, The C–O distance increases by 0.13 Ǻ, as the transformation from double to single bond occurs. Similarly, the C–N bond distance decreases by about 0.10 Ǻ, as it goes from a single to a double bond. The distance between the hydrogen atom to be transferred and the oxygen atom to receive it goes from 2.46 Å in EA, to 1.35 Å in TS1, and 0.97 Å in EI. Minor variations in the bond angles are also observed in the EA ⇆ EI process. Similar observations can be made for the ZA ⇆ EI and ZA ⇆ ZI processes.
When the complex between E-Amide and E-Imide with an explicit solvent molecule in the EAW ⇆ EIW process is calculated in aqueous solution, small geometrical variations of the E-Amide and E-Imide molecules take place. However, the CNH angle of the E-Amide form increases from 116° to 127° when the water complex is formed to facilitate the double hydrogen-bond with the assisting water molecule. When focusing on EAW, a hydrogen atom of the water molecule is located 1.93 Å away from the carbonyl oxygen and the oxygen atom is 2.13 Å away from the amidic hydrogen. These distances become 1.22 and 1.22 Å in TS2, respectively. In EIW, the oxygen atom of the solvent molecule is 1.78 Å from the hydroxyl hydrogen and a hydrogen atom is 2.18 Å away from the nitrogen atom. Similar comments can be made for the ZAW ⇆ ZIW process, but in this case, the water molecule assists the transfer of the amidic hydrogen to the hydroxide oxygen during the second step of the mechanism.
For the EA ⇆ EI process, the maximum on the potential of mean force (PMF) curve in the SMD simulation was reached at a N–H distance of 1.30 Å and at an angle with the oxygen atom of 102.6°, in agreement with the MP2-PCM optimized geometry of TS1 (see Fig. 4). For the water-assisted mechanism EAW ⇆ EIW, the maximum on the PMF curve corresponds to the TS2 structure reported in Fig. 4. Similarly, the location of transition states in the PMF curves for the processes that start with Z-Amide as reactant (TS3 through TS7) are in good agreement with the quantum geometries shown in Fig. 4.
3.3. Atomic charges and solvation
Analysis of the charges on the atoms involved in the amide-imide intramolecular proton transfers passing through different TSs, listed in Fig. 3 and 4, shows that there is a progressive decrease of the charge on the carbonyl oxygen of the amides as a result of the formation of the new O–H bond and the transformation from double to single bond. The zwitterion intermediates, Z± and ZW±, show an increase of the negative charge on the (previous) hydroxyl oxygen as a result of the first intramolecular proton transfer from ZA and ZAW, respectively. When an explicit water molecule participates in the reaction mechanism, an increase in the charge on the hydrogen atom of water which is hydrogen-bonded to either EAW or ZAW is also observed.Table 1 displays the average number of water molecules in the first hydration shell (solvent molecules that are within 2.5 Å of the solute) of each species considered. These values were obtained by performing 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps of traditional MD simulation for each rigid structure of solute. The MP2-PCM and M06-2X-SMD Gibbs free energies of solvation (ΔGsolv) are also shown in Table 1. The values reported in this table are intimately related to the structure and atomic charges of the species considered.
000 steps of traditional MD simulation for each rigid structure of solute. The MP2-PCM and M06-2X-SMD Gibbs free energies of solvation (ΔGsolv) are also shown in Table 1. The values reported in this table are intimately related to the structure and atomic charges of the species considered.
| Species | EA | TS1 | EI | EAW | TS2 | EIW | ZA | TS3 | TS4 |
|---|---|---|---|---|---|---|---|---|---|
| a ΔGsolv = Gsolution − Ggas.b Average number of water molecules on the first solvation shell.c MP2-PCM/6-311++G(d,p).d M06-2X-SMD/6-311++G(d,p). | |||||||||
| 〈n〉b | 4.4 | 4.2 | 4.0 | 5.3 | 5.2 | 4.8 | 5.1 | 4.6 | 4.9 |
| ΔGsolvc | −6.9 | −5.6 | −4.7 | −8.5 | −8.2 | −7.6 | −8.6 | −6.8 | −9.0 |
| ΔGsolvd | −11.9 | −9.5 | −8.1 | −15.6 | −14.8 | −11.8 | −14.6 | −10.6 | −13.1 |
The results reported in Table 1 allow us to conclude that: (a) amides (EA and ZA) are more hydrated than their corresponding imide forms (EI and ZI), because the carbonyl oxygen in the amides has the highest affinity for solvent molecules: the same applies when comparing the hydration of their water complexes (EAW and ZAW are more hydrated than EIW and ZIW, respectively); (b) due to an increase in the sites that can be solvated, the water complexes (EAW, EIW, ZAW and ZIW) are more hydrated than their corresponding simpler systems (EA, EI, ZA and ZI); (c) the hydration of Z-isomers (ZA, ZAW and ZIW) is greater than that of E-isomers (EA, EAW and EIW), respectively; (d) the degree of hydration of the TS of the elementary amide-imide transformations is in between the hydration of the corresponding reactant and product; (e) the protonation of the carbonyl oxygen in ZA and ZAW to form Z± and ZW±, respectively, reduces the hydration of these species on this atom, but increases on the other oxygen atom: hence the solvation energy increases relative to both the reactant and product in each case (Z± and ZW± are more hydrated than their corresponding species: ZA, ZI, ZAW and ZIW); (f) although the MP2-PCM and M06-2X-SMD ΔGsolv values show similar trends, the M06-2X-SMD values are greater than the MP2-PCM values for each species.
Fig. 5 shows a schematic representation of the hydration around the species involved in the unassisted E-Amide ⇆ E-Imide tautomerization. The water molecules marked with a circle represent those which are hydrogen-bonded to the solute (solvent molecules that are within 2.1 Å of the solute). In the first solvation layer of EA there are five molecules of water; three around the carbonyl oxygen, one around of the hydroxyl group and one near the amidic hydrogen. The first solvation layer of EI contains two water molecules near each hydroxyl group. The number of solvent molecules in the first solvation layer of TS1 is similar to that of EI, but in TS1 there are no solvent molecules where the proton transfer occurs. Similar descriptions can be made for other mechanisms.
 | ||
| Fig. 5 Hydration for the EA ⇆ EI mechanism from MD simulations. | ||
3.4. Kinetic and thermodynamic calculations
The calculated thermodynamic data (standard enthalpies and Gibbs free energies of formation) from electronic structure calculations in the gas phase and in solution at 298.15 K for each of the stationary points considered in this study are listed in Table S1 of the ESI† section. Table 2 displays the Gibbs free energy changes along the reaction profile of the five amide-imide transformations studied (see Section 3.1. and Fig. 2) from electronic structure calculations, in the gas phase (MP2 and M06-2X) and in solution (QCISD-PCM//MP2-PCM, MP2-PCM and M06-2X-SMD), and from SMD simulations. Previously reported values (calculated or experimental) are shown as well. The equivalent enthalpy changes (enthalpies of reaction and enthalpies of activation) from electronic calculations in solution are displayed in Table S2.† The calculated equilibrium constants (K) in the gas phase and in solution for each of the Amide ⇆ Imide elementary steps and global processes studied are reported in Table 3. The K values for the reverse processes, Imide ⇆ Amide, are displayed in Table S3.† Rate constants for the forward and reverse elementary steps calculated in solution at the QCISD-PCM//MP2-PCM, MP2-PCM and M06-2X-SMD levels of theory are shown in Table 4. Table S4† displays the calculated values for the tunneling factors for each elementary step in which a hydrogen ion is transferred, as well as the data used to obtain these values.| MP2 | M06-2X | QCISD-PCM//MP2-PCM | MP2-PCM | M06-2X-SMD | SMD | Other studies | ||
|---|---|---|---|---|---|---|---|---|
| a Ref. 15 (gas, 0 K).b Ref. 13 (gas).c Ref. 13 (exp, gas).d Ref. 11.e Ref. 12.f Ref. 10 (gas). | ||||||||
| (a) | EA | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| TS1 | 39.1 | 40.5 | 42.0 | 40.5 | 43.0 | 44.6 | 40.8a | |
| EI | 3.2 | 2.7 | 5.2 | 5.4 | 6.5 | 7.7 | 2.8b,c,3.9d,e, 4.7a | |
| (b) | EAW | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| TS2 | 18.2 | 17.5 | 19.3 | 18.5 | 18.2 | 23.3 | ||
| EIW | 2.6 | 2.1 | 2.9 | 3.5 | 5.9 | 2.4 | 4.2e,f | |
| (c) | ZA | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| TS3 | 12.6 | 12.4 | 12.2 | 14.5 | 16.5 | 16.1 | ||
| EA | −0.5 | −1.5 | −0.3 | 1.2 | 1.2 | 2.5 | −1.2d,e,1.4a, 1.8c | |
| TS1 | 38.6 | 39.0 | 41.7 | 41.7 | 44.2 | 47.1 | ||
| EI | 2.7 | 1.2 | 4.9 | 6.6 | 7.7 | 10.2 | 4.7c, 5.4d,e, 6.2a | |
| (d) | ZA | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| TS4 | 12.5 | 10.6 | 13.4 | 12.1 | 12.2 | 16.0 | ||
| Z± | 13.5 | 12.4 | 14.6 | 12.7 | 11.1 | 13.0 | ||
| TS5 | 50.7 | 50.1 | 55.6 | 53.7 | 55.7 | 65.8 | ||
| ZI | −0.5 | −2.0 | 2.0 | 3.5 | 5.4 | 5.7 | 0.8d,e,2.2c, 2.7f, 3.8a | |
| (e) | ZAW | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | 0.0 | |
| TS6 | 11.9 | 14.2 | 12.6 | 10.7 | 10.9 | 14.4 | ||
| ZW± | 12.3 | 16.8 | 12.7 | 10.7 | 10.0 | 11.5 | ||
| TS7 | 27.2 | 31.7 | 30.8 | 29.8 | 29.0 | 38.7 | ||
| ZIW | −1.4 | 1.0 | 0.0 | 2.1 | 3.6 | 3.8 | 0.6e | |
| MP2 | M06-2X | QCISD-PCM//MP2-PCM | MP2-PCM | M06-2X-SMD | SMD | ||
|---|---|---|---|---|---|---|---|
| (a) | EA ⇆ EI (TS1) | 4.4 × 10−3 | 1.1 × 10−2 | 1.5 × 10−4 | 1.1 × 10−4 | 1.8 × 10−5 | 2.3 × 10−6 |
| (b) | EAW ⇆ EIW (TS2) | 1.2 × 10−2 | 3.1 × 10−2 | 7.4 × 10−3 | 2.6 × 10−3 | 5.0 × 10−5 | 1.7 × 10−2 |
| (c) | ZA ⇆ EA (TS3) | 2.4 | 13 | 1.6 | 0.14 | 0.12 | 1.5 × 10−2 |
| ZA ⇆ EI | 1.1 × 10−2 | 0.13 | 2.4 × 10−4 | 1.6 × 10−5 | 2.2 × 10−6 | 3.3 × 10−8 | |
| ZAW ⇆ EIW | 0.29 | 2.7 × 10−2 | 6.0 × 10−2 | 1.4 × 10−4 | 8.2 × 10−6 | ||
| (d) | ZA ⇆ Z± (TS4) | 1.4 × 10−10 | 8.1 × 10−10 | 1.9 × 10−11 | 4.6 × 10−10 | 7.3 × 10−9 | 3.0 × 10−10 |
| Z± ⇆ ZI (TS5) | 1.7 × 1010 | 36 × 1010 | 1.8 × 109 | 5.9 × 106 | 1.5 × 104 | 2.2 × 105 | |
| ZA ⇆ ZI | 2.3 | 30 | 3.3 × 10−2 | 2.7 × 10−3 | 1.1 × 10−4 | 6.6 × 10−5 | |
| (e) | ZAW ⇆ ZW± (TS6) | 9.8 × 10−8 | 5.1 × 10−13 | 4.8 × 10−10 | 1.5 × 10−8 | 4.3 × 10−8 | 3.7 × 10−9 |
| ZW± ⇆ ZIW (TS7) | 1.1 × 108 | 3.4 × 1011 | 2.1 × 109 | 2.0 × 106 | 5.2 × 104 | 4.4 × 105 | |
| ZAW ⇆ ZIW | 10 | 0.17 | 1.0 | 3.0 × 10−2 | 2.2 × 10−3 | 1.6 × 10−3 | |
| kfwd (s−1) | krev (s−1) | ||||||
|---|---|---|---|---|---|---|---|
| QCISD-PCM//MP2-PCM | MP2-PCM | M06-2X-SMD | QCISD-PCM//MP2-PCM | MP2-PCM | M06-2X-SMD | ||
| a Effective rate constants calculated as keff = k2K1 using the pre-equilibrium approximation (see details in Section S2). | |||||||
| (a) | EA → EI (TS1) | 6.4 × 10−15 | 6.03 × 10−14 | 2.52 × 10−15 | 4.4 × 10−11 | 5.45 × 10−10 | 1.42 × 10−10 |
| (b) | EAW → EIW (TS2) | 2.0 | 6.26 | 1.07 | 2.7 × 102 | 2.45 × 103 | 2.13 × 104 |
| (c) | ZA → EA (TS3) | 7.7 × 103 | 141 | 4.87 | 4.7 × 103 | 992 | 39.9 |
| ZA → EIa | 1.02 × 10−14 | 8.44 × 10−15 | |||||
| (d) | ZA → Z± (TS4) | 1.0 × 103 | 9.11 × 103 | 1.55 × 104 | 17 | 2.00 × 1013 | 2.13 × 1012 |
| Z± → ZI (TS5) | 5.3 × 10−28 | 1.24 × 10−23 | 2.29 × 10−24 | 3.0 × 10−37 | 2.10 × 10−30 | 1.49 × 10−28 | |
| ZA → ZIa | 1.01 × 10−38 | 5.70 × 10−33 | |||||
| (e) | ZAW → ZW± (TS6) | 3.6 × 103 | 9.22 × 104 | 2.11 × 105 | 7.5 × 1012 | 6.02 × 1012 | 4.87 × 1012 |
| ZW± → ZIW (TS7) | 1.3 × 10−9 | 6.56 × 10−9 | 6.64 × 10−9 | 5.9 × 10−19 | 3.34 × 10−15 | 1.29 × 10−13 | |
| ZAW → ZIWa | 6.24 × 10−19 | 9.84 × 10−17 | |||||
From a qualitative point of view, the three sets of electronic structure calculations in solution are very similar for each of the five mechanisms studied. The M06-2X relative G values (which are the standard Gibbs free energies of activation, ΔG≠, or reaction, ΔG) are usually slightly larger than the MP2 values, with 2.5 kcal mol−1 the largest difference found. The differences between the QCISD//MP2 and MP2 relative G values are 0.2 kcal mol−1 on average (1.4 kcal mol−1 is the mean absolute difference), with 2.4 kcal mol−1 the largest difference found.
Even though the Gibbs free energies of formation in the gas phase are greater than in solution, with differences of 4.6–13.8 kcal mol−1 for the MP2 case and 7.2–23.3 kcal mol−1 for the M06-2X case (see Table 1), the ΔG≠ and ΔG values in solution are usually larger than those in the gas phase at both levels of theory (see Table 2). The discussion that follows will focus on the results obtained in aqueous solution.
In general, there is good qualitative agreement between the values predicted from the SMD simulations and from the electronic structure calculations. However, the SMD relative G values are in most cases greater than each of the calculated values with the electronic structure methods; hence for both thermodynamic and kinetic results, the SMD values are in closer agreement with the M06-2X results. With the exception of the values for TS5 and TS7, the relative G differences between SMD and MP2-PCM (M06-2X-SMD) are in the 0.3–5.4 (0.2–5.1) kcal mol−1 range. In the SMD simulations, a semi-empirical functional is used (DFTB) and the solvent is treated as a discrete medium, not as a continuum (which is the case of the solvent methods applied with the electronic structure calculations). Even when the solvent molecules are treated classically, explicit solute–solvent interactions are now accounted for, in addition to the explicit interactions present in the case of the water-assisted mechanisms. As a consequence, the processes considered tend to be more energetic when calculated from the SMD simulations, hence larger ΔG≠ values are calculated in most cases. Due to a significant charge separation, this situation might play a greater role when proton transfer barriers are calculated from the zwitterions (Z± and ZW±) leading to TS5 and TS7.
The elementary and global processes studied for each of the five Amide ⇆ Imide tautomerizations in aqueous solution are slightly endothermic (ΔH°> 0) and endergonic (ΔG° > 0), except the elementary steps in which the zwitterion intermediate (Z± or ZW±) is the reactant. These two steps are calculated to be exothermic and thermodynamically spontaneous at room temperature (with equilibrium constants K > 1, see Table 3). When considering the MP2 calculations, the elementary and global processes (except ZA → Z±) are more thermodynamically favoured (have a larger K) in the gas phase than in solution. All water-assisted processes are more thermodynamically favoured (having a smaller ΔG° or larger K value) than the corresponding unassisted ones. From a thermodynamic point of view, the Z-Amide → Z-Imide process seems to be slightly more favoured than all other processes considered.
Unless otherwise indicated, the discussion that follows uses the values obtained from the electronic structure calculations (following the order QCISD-PCM//MP2-PCM value, MP2-PCM value, M06-2X-SMD value). The calculated ΔG for the EA ⇆ EI process via the non-assisted mechanism (5.2, 5.4, 6.5 kcal mol−1) is slightly greater than the value reported by other authors (ΔG = 2.8–4.7 kcal mol−1). However, our calculated ΔG≠ values (42.0, 40.5, 43.0 kcal mol−1) are in good agreement with the previously reported one (40.8 kcal mol−15). When the water-assisted mechanism EAW ⇆ EIW, in which the explicit solvent molecule facilitates the proton transfer is considered, a significant reduction (42–46%) of the calculated ΔG≠ value occurs (19.3, 18.5, 18.2 kcal mol−1). These results are in agreement with the 50% reduction previously reported.10 The calculated rate constant (see Table 4) increases ca. 1014 times going from 6.4 × 10−15 s−1 to 2.0 s−1 (QCISD-PCM//MP2-PCM results) when the water-assisted process is considered. This significant rate constant increase (and ΔG≠ reduction) can be partly justified from a structural point of view since the explicit presence of water reduces the strain in the four-member ring in TS1, relative to the five-member ring in TS2. However, a small decrease in ΔG is observed with each method applied (2.9, 3.5, 5.9 kcal mol−1). The potential of mean force (PMF) curves of the elementary processes from SMD simulations, which reflect the previous comments, are displayed in Fig. 6.
 | ||
| Fig. 6 PMF of the elementary processes (a) EA ⇆ EI (—) and (b) EAW ⇆ EIW (…) from SMD simulations. | ||
The three tautomeric processes studied that start from the Z-Amide form are complex; they occur in two kinetic steps. The PMF curves of these processes from SMD simulations are displayed in Fig. 7. For the ZA ⇆ EI process, the ΔG≠ and ΔG values for the internal rotation to form EA were calculated to be 12.2, 14.5, 16.5 kcal mol−1 and −0.3, 1.2, 1.2 kcal mol−1, respectively. The global ΔG for the ZA ⇆ EI process was calculated to be 4.9, 6.6, 7.7 kcal mol−1.
 | ||
Fig. 7 PMF of the complex processes (c) ZA ⇆ EI (![[dash dot, graph caption]](https://www.rsc.org/images/entities/char_e090.gif) ), (d) ZA ⇆ ZI (—) and (e) ZAW → ZIW (…) from SMD simulations. ), (d) ZA ⇆ ZI (—) and (e) ZAW → ZIW (…) from SMD simulations. | ||
For the Z-Amide to Z-Imide transformation, as previously observed for the corresponding E-isomers, the explicit participation of a solvent molecule also produces a decrease in the calculated ΔG≠ of each of the two steps involved, but the reduction is much more significant for the second (rate-determining) step. For the first step, in which a proton is transferred between the two oxygen atoms and a zwitterion is formed, the calculated ΔG≠ goes from 13.4, 12.1, 12.2 kcal mol−1 to 12.6, 10.7, 10.9 kcal mol−1. The ΔG≠ of the second proton transfer, which for mechanism (e) (see Fig. 2) involves the explicit solvent molecule, goes from 55.6, 53.7, 55.7 kcal mol−1 to 30.8, 29.8, 29.0 kcal mol−1 for reduction which is ca. 50%, similar to the result found for the E-Amide ⇆ E-Imide process.
The additional stabilization due to solvation associated with internal charge separation in the two zwitterion structures, Z± and ZW± (see Table 1), relative to that of the TSs that form from them (TS5 and TS7, respectively), contributes to the much larger ΔG≠ of the second step of mechanisms (d) and (e). Structural aspects once again help us to rationalize the reduction in ΔG≠ when comparing the two TSs of mechanisms (d) and (e). The explicit solvent molecule makes the structure of ZAW closer to that of TS6, relative to a similar comparison between ZA and TS4. Similarly, the explicit presence of water reduces the strain in the three-member ring in TS5, relative to the five-member ring in TS7. In both cases, the proton transfer is favoured for the mechanism with the explicit solvent molecule. The explicit solute–solvent interactions accounted for in the SMD simulations might be the reason for the much greater ΔG≠ values calculated for these four TSs, which differ from the electronic structure calculation values by 1.8–10.2, 3.7–12.1 and 3.5–10.1 kcal mol−1.
Given that the complex processes (d) and (e) have an initial fast step, followed by an irreversible rate-determining step, the pre-equilibrium approximation can be applied to calculate an effective rate constant (keff = k2K1). Using the QCISD-PCM//MP2-PCM values, keff is 1.0 × 10−38 s−1 for the unassisted process and increases ca. 1019 times (keff = 6.2 × 10−19 s−1) when considering the water-assisted process. Note that in both cases, the calculated keff values are significantly smaller than the corresponding calculated rate constants for the E-Amide → E-Imide processes.
Similar to what was previously observed with the E-isomers, a small decrease in the Gibbs free energy of reaction is calculated with each method applied when comparing the unassisted and water-assisted mechanisms (see Fig. 2d and e). The ΔG of the first step goes from 14.6, 12.7, 11.1 kcal mol−1 to 12.7, 10.7, 10.0 kcal mol−1, while for the global process the reduction in ΔG goes from 2.0, 3.5, 5.4 kcal mol−1 to 0.0, 2.1, 3.6 kcal mol−1. The calculated global ΔGZA→ZI values are in good agreement with values previously reported by other authors (ΔGZA→ZI = 0.8–3.8 kcal mol−1 (ref. 10–15)) and the experimental gas-phase value of 2.2 kcal mol−1.13
When focusing on the two Z-Amide transformations without water assistance, to E-Imide (c) and to Z-Imide (d), the first process (with QCISD-PCM//MP2-PCM barriers of 12.2 and 41.7 kcal mol−1; keff = 1.02 × 10−14 s−1) is more kinetically favoured than the Z-Amide → Z-Imide process (with QCISD-PCM//MP2-PCM barriers of 13.4 and 55.6 kcal mol−1 relative to ZA; keff = 1.0 × 10−38 s−1). This comparison does not change when considering the water-assisted mechanisms. The ZAW → EIW transformation has an initial QCISD-PCM//MP2-PCM barrier that should be greater than 12.2 kcal mol−1 (the water molecule in ZAW would hinder the internal rotation around the C–N single bond to form EAW, relative to the value calculated for the ZA → EA internal rotation), and a second barrier of 18.8 kcal mol−1,*1corresponding to the EAW → EIW tautomerization. Since these two barriers would likely be somehow similar, there would not be a well-defined rate-determining step (RDS) for this mechanism. The ZAW → ZIW transformation has an initial QCISD-PCM//MP2-PCM barrier of 12.6 kcal mol−1 to form the zwitterion intermediate ZW±, followed by the RDS with a barrier of 30.8 kcal mol−1, relative to ZAW. Hence, from a kinetic point of view, the most favoured tautomeric processes from Z-Amide is Z-Amide → E-Imide. Overall, the tautomeric conversion which is the most kinetically favoured is E-Amide → E-Imide.
4. Conclusions
The thermodynamic and kinetic study of the different tautomeric equilibria of acetohydroxamic acid in aqueous solution have been studied making use of three electronic structure methods (QCISD//MP2, MP2 and M06-2X) and SMD simulations. Mechanisms have been calculated with and without the presence of an explicit solvent molecule. From a thermodynamic point of view, Z-Amide is the most favourable species, followed by E-Amide. The elementary E-Amide → E-Imide process is the most kinetically favoured. The tautomeric processes from Z-Amide are complex and its conversion to E-Imide is the most kinetically favoured. In all cases, the explicit consideration of a solvent molecule (in addition to accounting for other solvent effects depending on the methodology applied) led to significantly smaller Gibbs free energies of activation for the rate-determining step, and slightly smaller Gibbs free energies of reaction.Consistent results were obtained with the four methodologies applied, which highlights the validity of SMD simulations to study processes where proton transfers occur. To the best of our knowledge, this is the first study in which MD simulations are applied to study the Amide-Imide tautomerism, and the first complete study of the kinetics and the thermodynamics of the tautomerization processes in acetohydroxamic acid.
Acknowledgements
This research was sponsored by the Consejería de Infraestructuras y Desarrollo Tecnológico de la Junta de Extremadura (Project GRU10036) and the Natural Sciences and Engineering Research Council of Canada (NSERC). The authors wish to thank Prof. Raul Alvarez-Idaboy for fruitful discussions.References
- C. J. Cramer and D. G. Truhlar, Structure and Reactivity in Aqueous Solution: Characterization of Chemical and Biological Systems, Americal Chemical Society, Washington D.C, 1994 Search PubMed.
- A. Warshel, Computer Modeling of Chemical Reactions in Enzymes and Solutions, Wiley & Sons, New York, 1991 Search PubMed.
- A. Muller, H. Ratjczak and W. Junge, Electron and Proton Transfer in Chemistry and Biology, Elsevier, Amsterdam, 1992 Search PubMed.
- P. Bala, P. Grocowski, B. Lesyg and J. A. McCammon, Quantum Mechanical Simulation Methods for Studying Biological Systems, Springer, Berlin, 1995 Search PubMed.
- J. Aqvist, Modelling of Proton Transfer Reactions in Enzymes, in Computational Approaches to Biochemical Reactivity, ed. G. Naray-Szabó and A. Warshel, Kluwer Academic Publisher, Netherlands, 1997, pp. 341–362 Search PubMed.
- K. Binder, G. Ciccotti and M. Ferrario, Computer Simulations in Condensed Matter Systems: From Materials to Chemical Biology, Springer, Berlin, 2006, vol. 2 Search PubMed.
- G. Náray-Szabó and A. Warshel, Computational Approaches to Biochemical Reactivity, Springer, Berlin, 2002 Search PubMed.
- J. C. Kotz, P. M. Treichel and J. Townsend, Chemistry and chemical reactivity, Thomsom Higher Education, Belmont, 2009 Search PubMed.
- O. Tapia and J. Bertran, Solvent Effects and Chemical Reactivity, Springer, Berlin, 2003 Search PubMed.
- H. Tavakol, J. Mol. Struct.: THEOCHEM, 2009, 916, 172–179 CrossRef CAS PubMed.
- M. L. Senent, A. Niño, C. Muñoz Caro, S. Ibeas, B. García, J. M. Leal, F. Secco and M. Venturini, J. Org. Chem., 2003, 68, 6535–6542 CrossRef CAS PubMed.
- N. Mora-Diez, M. L. Senent and B. García, Chem. Phys., 2006, 324, 350–358 CrossRef CAS PubMed.
- M. Saldyka and Z. Mielke, Vib. Spectrosc., 2007, 45, 46–54 CrossRef CAS PubMed.
- N. J. Fitzpatrick and R. Mageswaran, Polyhedron, 1989, 8, 2255–2263 CrossRef CAS.
- R. Kakkar, R. Grower and P. Chadha, Org. Biolo. Chem., 2003, 1, 2200–2206 RSC.
- D. H. Wu and J. J. Ho, J. Phys. Chem., 1998, 102, 3582–3586 CrossRef CAS.
- J. Yazal and Y. P. Pang, J. Phys. Chem. A, 1999, 103, 8346–8350 CrossRef.
- L. J. Yamin, C. A. Ponce, M. R. Estrada and F. T. Vert, J. Mol. Struct.: THEOCHEM, 1996, 360, 109–117 CrossRef CAS.
- D. M. Stinchcomb and J. Pranata, J. Mol. Struct.: THEOCHEM, 1996, 370, 25–32 CrossRef CAS.
- B. Garcia, S. Ibeas, J. M. Leal, M. L. Senent, A. Niño and C. Muñoz-Caro, Chem.–Eur. J., 2000, 6(14), 2644–2652 CrossRef CAS.
- C. Muñoz Caro, A. Niño, M. L. Senent, S. Ibeas and J. M. Leal, J. Org. Chem., 2000, 65, 405 CrossRef PubMed.
- M. Saldyka and Z. Mielke, Pol. J. Chem., 2003, 77(11), 1587–1598 CAS.
- A. Kaczor and L. M. Proniewicz, J. Mol. Struct., 2004, 704, 189–196 CrossRef CAS PubMed.
- M. Saldyka and Z. Mielke, Spectrochim. Acta, Part A, 2005, 61(7), 1491–1497 CrossRef PubMed.
- S. P. Gupta, Hydroxamic acids. A unique family of chemical with multiple biological activities, Springer, Germany, 2013 Search PubMed.
- D. A. Brown, L. P. Cuffe, M. Geraldine, N. Fitzpatrick, W. K. Glass and K. M. Herlihy, Collect. Czech. Chem. Commun., 2001, 66, 99–108 CrossRef CAS.
- A. Artmenko, E. K. Anufriev, I. V. Tikunova and O. Ekner, J. Appl. Spectrosc., 1980, 33, 758–761 CrossRef.
- R. Kakkar, R. Grover and P. Chadha, Org. Biomol. Chem., 2003, 1, 2200–2206 CAS.
- L. Turi, J. J. Dannenberg, J. Rama and O. N. Ventura, J. Phys. Chem., 1992, 96(9), 3709–3712 CrossRef CAS.
- T. V. Surova and A. N. Enyashin, J. Struct. Chem., 2003, 44(2), 297 CrossRef CAS.
- M. Saldyka and Z. Mielke, Chem. Phys. Lett., 2003, 371(5), 713–718 CrossRef CAS.
- O. N. Ventura, J. B. Rama, L. Turi and J. J. Dannenberg, J. Am. Chem. Soc., 1993, 115(3), 5754–5761 CrossRef CAS.
- S. R. Emamian, L. R. Domingo and S. F. Tayyari, J. Mol. Graphics Modell., 2014, 49, 47–54 CrossRef CAS PubMed.
- S. Tolosa, A. Hidalgo and J. A. Sansón, J. Phys. Chem. B, 2012, 116(43), 13033–13044 CrossRef CAS PubMed.
- S. Tolosa, J. A. Sansón and A. Hidalgo, J. Phys. Chem. A, 2007, 111, 339–344 CrossRef PubMed.
- S. Tolosa, J. C. Corchado, A. Hidalgo and J. A. Sansón, J. Phys. Chem. A, 2007, 111, 13515–13520 CrossRef PubMed.
- S. Tolosa, A. Hidalgo and J. A. Sansón, J. Phys. Chem. A, 2009, 113, 1858–1863 CrossRef PubMed.
- S. Tolosa, A. Hidalgo and J. A. Sansón, Theor. Chem. Acc., 2010, 127, 671–679 CrossRef.
- S. Tolosa, A. Hidalgo and J. A. Sansón, J. Struct. Chem., 2011, 22, 909–915 CrossRef.
- S. Tolosa, A. Hidalgo and J. A. Sansón, J. Mol. Model., 2014, 20(3), 2147 CrossRef PubMed.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Mez, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, J. Am. Chem. Soc., 1995, 117, 5179–5197 CrossRef CAS.
- W. Damm, A. Frontera, J. Tirado-Rives and W. L. Jorgensen, J. Comput. Chem., 1997, 18, 1955 CrossRef CAS; W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, J. Am. Chem. Soc., 1996, 118, 11225 CrossRef; W. L. Jorgensen and J. Tirado-Rives, J. Am. Chem. Soc., 1988, 110, 1657 CrossRef; G. Kaminski, E. M. Duffy, T. Matsui and W. L. Jorgensen, J. Phys. Chem., 1994, 98, 13077 CrossRef.
- S. Izrailev, S. Stepaniants, B. Isralewitz, D. Kosztin, H. Lu, F. Molnar, W. Wriggers and K. Schulten, Steered Molecular Dynamics, in Computational Molecular Dynamics, Challenges, Methods, Ideas of Lectures Notes in Computational Science and Engineering, ed. P. Deuflhard, J. Hermans, B. Leimkuhler, A. E. Mark, S. Reich and R. D. Skell, Springer-Verlag, Berlin, 1998, vol. 4, pp. 39–65 Search PubMed.
- B. Isralewitz, M. Gao and K. Schulten, Curr. Opin. Struct. Biol., 2001, 11, 224–230 CrossRef CAS.
- A. Krammer, H. Lu, B. Isralewitz, K. Schulten and V. Vogel, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 1351–1356 CrossRef CAS.
- M. Gao, D. Craig, V. Vogel and K. Schulten, J. Mol. Biol., 2002, 323, 939–950 CrossRef CAS.
- S. Izrailev, S. Stepaniants, M. Balsera, Y. Oono and H. Schulten, Biophys. J., 1997, 72, 1568–1581 CrossRef CAS.
- M. V. Bayas, K. Schulten and D. Leckband, Biophys. J., 2003, 84, 2223–2233 CrossRef CAS.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision A.1., Gaussian, Wallingford, CT, 2009 Search PubMed.
- S. Miertus, E. Scrocco and J. Tomasi, Chem. Phys., 1981, 55, 117–129 CrossRef CAS.
- Y. Zhao and D. G. Truhlar, Theor. Chem. Acc., 2008, 120, 215–241 CrossRef CAS.
- (a) A. V. Marenich, C. J. Cramer and D. G. Truhlar, J. Phys. Chem. B, 2009, 113, 6378 CrossRef CAS PubMed; (b) C. J. Cramer and D. G. Truhlar, SMx Continuum Models for Condensed Phases, in Trends and Perspectives in Modern Computational Science; Lecture Series on Computer and Computational Sciences, ed. G. Maroulis and T. E. Simos, Brill/VSP, Leiden, 2006, vol. 6. pp. 112–140 Search PubMed.
- R. S. Mulliken, J. Chem. Phys., 1995, 23(10), 1833–1840 CrossRef PubMed.
- C. Jarzynski, Phys. Rev. Lett., 1997, 78, 2690–2693 CrossRef CAS.
- D. A. Case, T. A. Darden, I. T. E. Cheatham III, C. L. Simmerling, J. Wang, R. E. Duke, R. Luo, R. C. Walker, W. Zhang, K. M. Mert, B. Roberts, S. Hayik, A. Roitberg, G. Seabra, J. Swails, A. W. Götz, I. Kolossváry, K. F. Wong, F. Paesani, J. Vanicek, R. M. Wolf, J. Liu, X. Wu, S. R. Brozell, T. Steinbrecher, H. Gohlke, Q. Cai, X. Ye, J. Wang, M. J. Hsieh, G. Cui, D. R. Roe, D. H. Mathews, M. G. Seetin, R. Salomon-Ferrer, C. Sagui, V. Babin, T. Luchko, S. Gusarov, A. Kovalenko and P. A. Kollman, AMBER 12, University of California, San Francisco, 2012 Search PubMed.
- P. Ewald, Ann. Phys., 1921, 64, 253–287 CrossRef.
- T. Frauenheim, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, A. Sieck and G. Seifert, Mater. Res. Soc. Symp. Proc., 1998, 491, 91 CrossRef CAS PubMed.
- M. Elstner, D. Porezag, G. Jungnickel, J. Elsner, M. Haugk, T. Frauenheim, S. Suhai and G. Seifert, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 7260 CrossRef CAS.
- G. M. Seabra, R. C. Walker, M. Elstner, D. A. Case and A. E. Roitberg, J. Phys. Chem., 2007, 111, 5655–5664 CrossRef PubMed.
- R. C. Walker, M. F. Crowley and D. A. Case, J. Comput. Chem., 2008, 29, 1019–1031 CrossRef CAS PubMed.
- (a) C. Eckart, Phys. Rev., 1930, 35, 1303 CrossRef CAS; (b) H. Shin, J. Chem. Phys., 1963, 39, 2934 CrossRef CAS PubMed.
- R. L. Brown, J. Res. Natl. Bur. Stand., 1981, 86, 357 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: The calculated thermodynamic data (standard enthalpies and Gibbs free energies of formation) from electronic structure calculations in the gas phase and in solution at 298.15 K for each of the stationary points considered in this study (Table S1); enthalpy changes along the reaction profile of the five transformations studied from electronic structure calculations in solution (Table S2); equilibrium constants (K) calculated in the gas phase and in solution for each of the Imide ⇆ Amide elementary steps and global processes studied at 298.15 K (Table S3); calculated tunneling factors (κ) and data used for these calculations at 298.15 K (Table S4); MP2-PCM/6-311++G(d,p) Cartesian coordinates of the optimized stationary points considered in this study (Section S1); details on the rate constant comparison for elementary and complex processes (Section S2). See DOI: 10.1039/c4ra06124a |
| This journal is © The Royal Society of Chemistry 2014 |