
Combined spectroscopy and cyclic voltammetry investigates the interaction between [(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl anticancer drug and human serum albumin
Shan Huangab,
Fawei Zhua,
Qi Xiao*abc,
Quan Zhoua,
Wei Su*a,
Hangna Qiua,
Baoqing Hub,
Jiarong Shenga and
Chusheng Huanga
aCollege of Chemistry and Materials Science, Guangxi Teachers Education University, Nanning 530001, P. R. China. E-mail: qi.xiao@whu.edu.cn, aaasuwei@yahoo.com.cn; Fax: +86 771 3908065; Tel: +86 771 3908065
bKey Laboratory of Beibu Gulf Environment Change and Resources Utilization (Guangxi Teachers Education University), Ministry of Education, China
cThe State Key Laboratory of Virology, Wuhan University, China
First published on 30th July 2014
Abstract
In this article, the interaction between [(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl anticancer drug and human serum albumin (HSA) was investigated systematically under physiological conditions by using some spectroscopic methods (UV-vis absorption spectroscopy, fluorescence spectroscopy, FT-IR spectroscopy, CD spectroscopy), mass spectroscopy and cyclic voltammetry. The experimental results indicated that this anticancer drug could quench the intrinsic fluorescence of HSA through static quenching mechanism. The Stern–Volmer quenching model has been successfully applied, and the Stern–Volmer quenching constants together with the modified Stern–Volmer quenching constants at different temperatures were also calculated. The corresponding thermodynamic parameters ΔH, ΔG and ΔS were also calculated. The binding of this anticancer drug and HSA resulted in the formation of drug–HSA complex, and the electrostatic interaction played a major role in the complex stabilization. The distance r between the donor (HSA) and the acceptor (drug) was obtained through fluorescence resonance energy transfer theory. Competitive experiments indicated that the binding site of this anticancer drug to HSA was located at site I. The results of synchronous fluorescence spectra, three-dimensional fluorescence spectra, FT-IR spectra and CD spectra indicated that the microenvironment and the conformation of HSA were changed noticeably due to the presence of this anticancer drug. The results of mass spectra and cyclic voltammetry further confirmed the interaction between HSA and this anticancer drug. These results indicated that the biological activity of HSA was dramatically affected by the [(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl anticancer drug.
1. Introduction
As a kind of organometallic compound, ruthenium(II) arene complexes of thiosemicarbazone (TSC) with the half-sandwich type structure have attracted great attention by biochemists because of their super-high antibacterial, antiviral and anticancer activities.1–4 Due to their higher anticancer activity and lower toxicity than platinum drugs, ruthenium(II) arene complexes of TSC anticancer drugs have been widely researched by chemists and medical scientists, and have progressed into potential anticancer drugs of the near future. Some research has demonstrated the cytotoxicity and the anticancer activity of these ruthenium(II) arene complexes of TSC anticancer drugs. Beckford et al. reported the first structurally characterized ruthenium arene complexes with TSC ligands and further confirmed their good cytotoxic profiles against different human cancer cell lines.5,6 Smith and co-workers demonstrated the cytotoxicity and antiparasitic activity of some mono- and dinuclear ruthenium(II)-arene complexes with TSCs derivatives,7,8 and Gambino et al. also demonstrated the cytotoxic activities of some organometallic binuclear TSC ruthenium arene complexes.9,10 More recently, Su's group have synthesized a series of ruthenium(II) arene complexes with TSC ligands and further investigated their anticancer activities.11,12 Their results indicated that one of the derivatives of ruthenium(II) arene complexes of TSC anticancer drugs, {[(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl} (structure shown in Fig. 1), was regarded as a potential new anticancer drug with an IC50 value of less than 3.4 × 10−5 mol L−1 for human cancer cell lines, which indicates its further wide application in biomedicine. | ||
| Fig. 1 Emission spectra of HSA in the presence of various concentrations of this anticancer drug (pH 7.4, T = 298 K, λex = 295 nm). The insert corresponds to the Stern–Volmer plot of HSA with the increasing concentration of this anticancer drug at 298 K. c (HSA) = 2.0 × 10−6 mol L−1; c (drug)/(10−6 mol L−1), 1–11: 0; 0.1; 0.2; 0.3; 0.4; 0.5; 0.6; 0.7; 0.8; 0.9; 1.0; curve L shows the emission spectrum of this anticancer drug only, c (drug) = 1.0 × 10−6 mol L−1. | ||
Since ruthenium(II) arene complexes of TSC anticancer drugs could intercalate into the structure of nucleic acids, which might inhibit the biological functions of nucleic acids and cause the death of cancer cells,13–15 DNA or RNA has been demonstrated as the biological targets of these ruthenium(II) arene complexes of TSC anticancer drugs. But beyond that, as the assured potential of ruthenium(II) arene complexes of TSC anticancer drugs in therapy, their absorption, distribution, metabolism and excretion properties in organism are still unknown right now. So, a deeper understanding of their distinct modes of action and improving their selectivity, along with broadening their biomedical application remain the focus of active research.
The investigation of the interactions between ruthenium(II) arene complexes of TSC anticancer drugs and some important biomolecules became an effective approach for better understanding the mechanism of their anticancer activities, as well as the targeted design and synthesis of new efficient anticancer drugs. However, the research of the interaction between ruthenium(II) arene complexes of TSC anticancer drugs and serum albumin are rarely involved. As a kind of important biomolecules and the principal extracellular soluble protein during the circulatory system, serum albumin, which contributes significantly to the transportation, distribution and metabolism of drugs, is usually chosen as the model protein for the investigation of the interaction between some exogenous ligands and important biomolecules.16–20 Herein, due to the well-known spatial structures of human serum albumin (HSA),21–23 HSA was usually used as a model protein for the research of the interactions between exogenous ligands and serum albumin, including the interactions of different drugs and HSA.24–26
In this article, the spectroscopic and electrochemical investigations of the interactions between [(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl anticancer drug and HSA under physiological condition was reported. The interactions of this anticancer drug and HSA were systematically investigated through multispectroscopic approaches and cyclic voltammetry. The intrinsic fluorescence of HSA was quenched dramatically by this anticancer drug by a concentration-dependent manner. The Stern–Volmer quenching constants were decreased with the increase of the temperature, which proved that this anticancer drug could quench the intrinsic fluorescence of HSA effectively through static quenching mechanism. Some thermodynamic parameters, such as ΔH, ΔG and ΔS, were also calculated from both the modified Stern–Volmer equation and the corresponding quenching constants. The distance between this anticancer drug and HSA was calculated through the fluorescence resonance energy transfer (FRET) theory, and the binding site of this anticancer drug to HSA was further investigated. In addition, the microenvironmental and the conformational changes of HSA induced by this anticancer drug were analyzed through the synchronous fluorescence spectroscopy, three-dimensional fluorescence spectroscopy, FT-IR spectroscopy and CD spectroscopy. The binding number and the interaction constant between this anticancer drug and HSA were confirmed by mass spectra and cyclic voltammetry, respectively. This research could provide valuable information of biological action of this anticancer drug in organism, and might serve as important strategy for pharmacological and toxicological research of novel ruthenium(II) arene complexes with TSC anticancer drugs.
2. Materials and methods
2.1 Apparatus
The fluorescence spectra and intensities were performed on Perkin-Elmer LS-55 luminescence spectrometer (PerkinElmer, Waltham, MA, USA) equipped with a 20 kW xenon discharge lamp as light source and a thermostatic bath (DKB-501S, Shanghai Jing Hong Laboratory Instrument Co. Ltd, China). The UV-vis absorption spectra were measured on a TU-1901 UV-vis spectrophotometer at room temperature (Beijing Purkinje General Instrument Co., Ltd., Beijing, China). Quartz cells (1 cm path-length) were used for all measurements. The FT-IR spectra were recorded at room temperature on Nicolet iS10 spectrometer (Thermo, USA) equipped with a zinc selenide (ZnSe) attenuated total reflection (ATR) accessory, a deuterated triglycine sulfate (DTGS) detector, and a KBr beam splitter. The CD spectra were recorded on Jasco J-810 automatic recording CD spectropolarimeter (Jasco, Tokyo, Japan) using a cuvette with 1.0 cm path length. The mass spectra were recorded on ACQUITY UPLC/XEVO G2 Q TOF mass spectrometer (Waters Corporation, Milford, USA) equipped with an electrospray ionization probe. The cyclic voltammetric experiments were performed on a CHI-660E electrochemical workstation (Chenhua Instruments Inc., Shanghai, China) with a conventional three-electrode electrochemical testing system. The working electrode was a bare gold electrode (2 mm diameter), whereas the Ag/AgCl electrode served as the reference electrode and a platinum wire was used as the counter electrode. All pH measurements were made with a basic pH meter PB-10 (Sartorius Scientific Instruments Co., Ltd., Beijing, China).2.2 Materials
The ruthenium(II) arene complexes with TSC [(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl anticancer drug was synthesized and characterized according to the literature reported before.11 The structure of this anticancer drug was shown in Fig. 1. The stock solution of 1.0 × 10−3 mol L−1 drug was prepared by dissolving its crystals in DMSO. HSA (Sigma, St. Louis, MO, USA) was dissolved in PBS (0.1 mol L−1, pH 7.4) and stored at 4 °C. The concentration of HSA was determined spectrophotometrically using an extinction coefficient ε280nm = 36![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 600 L mol−1 cm−1.27 The stock solutions (1.0 × 10−3 mol L−1) of Warfarin (Sigma, St. Louis, MO, USA) and ibuprofen (Sigma, St. Louis, MO, USA) were prepared by dissolving the accurately weighed appropriate amounts of each compound and dissolving in ultrapure water. Then, the solution was diluted to the required volume with ultrapure water in practical use. Ultrapure water with a resistivity of 18.2 MΩ cm was produced by passing through a RiOs 8 unit followed by a Millipore-Q Academic purification set (Millipore, Bedford, MA, USA) and used throughout the whole experiments. All other reagents were of analytical reagent grade and used as received without further purification.
600 L mol−1 cm−1.27 The stock solutions (1.0 × 10−3 mol L−1) of Warfarin (Sigma, St. Louis, MO, USA) and ibuprofen (Sigma, St. Louis, MO, USA) were prepared by dissolving the accurately weighed appropriate amounts of each compound and dissolving in ultrapure water. Then, the solution was diluted to the required volume with ultrapure water in practical use. Ultrapure water with a resistivity of 18.2 MΩ cm was produced by passing through a RiOs 8 unit followed by a Millipore-Q Academic purification set (Millipore, Bedford, MA, USA) and used throughout the whole experiments. All other reagents were of analytical reagent grade and used as received without further purification.
2.3 Fluorescence spectra measurements
Steady-state fluorescence spectra of HSA (2.0 × 10−6 mol L−1) and HSA (2.0 × 10−6 mol L−1) at the present of this anticancer drug (0–1.0 × 10−6 mol L−1, with an interval of 1.0 × 10−7 mol L−1) were recorded at 298 K, 304 K, 310 K in the wavelength range of 300–450 nm. The width of the excitation and the emission slit was set to 5.0 nm and 5.0 nm, and the excitation wavelength of 295 nm was chosen in the experiment to avoid inner filter effect. Titrations were performed manually by using trace syringes and each spectrum was the average of three scans.The binding site investigation experiments were performed by using different site-specific probes (warfarin and ibuprofen for site I and site II on HSA, respectively) through keeping the concentrations of both HSA and site-specific probe constant but varying the concentration of this anticancer drug alone. The steady-state fluorescence spectra of the system were recorded and the corresponding binding constants values of drug–HSA–probe system were evaluated, which could decide the binding site of this specific drug. The concentrations of the site-specific probes and HSA were all 2.0 × 10−6 mol L−1.
The effects of common ions on the binding constant were investigated by recording the steady-state fluorescence spectra of drug–HSA system in the presence of some common metal ions, for example, Ag+, K+, Na+, Cu2+, Zn2+, Ca2+, Mg2+, Ni2+, Co2+, NO3−, SO42− and Cl− at 298 K upon the excitation wavelength at 295 nm. The concentrations of the adopted metal ions and HSA were all 2.0 × 10−6 mol L−1.
The synchronous fluorescence spectra of HSA alone and the drug–HSA system were scanned from 280–330 nm and 300–380 nm at the constant Δλ (Δλ = λem − λex) of 15 nm and 60 nm, respectively. The Δλ of 15 nm and 60 nm reflected the spectrum characteristics of tyrosine (Tyr) and tryptophan (Trp) residues of HSA were observed. The concentration of HSA was kept at 2.0 × 10−6 mol L−1.
The three-dimensional fluorescence spectra of HSA alone and the drug–HSA system were performed with the emission wavelength range was 200–500 nm and the excitation wavelength range was 200–350 nm with increment of 5 nm. Other scanning parameters were just the same as that of the steady-state fluorescence spectra. The concentrations of HSA and anticancer drug were 2.0 × 10−6 mol L−1 and 1.0 × 10−6 mol L−1, respectively.
2.4 UV-vis absorbance measurements
The UV-vis absorption spectra of free HSA (2.0 × 10−6 mol L−1), anticancer drug alone (2.0 × 10−6 mol L−1) as well as the drug–HSA system (equal molar ratio) were measured from 200 nm to 350 nm. The solutions of the blank buffer and sample were placed in the reference and sample cuvettes, respectively.2.5 FT-IR spectra measurements
The FT-IR spectra of free HSA and drug–HSA system in PBS were recorded in the range of 1450–1800 cm−1 with 128 interferograms to ensure a good signal-to-noise ratio. The corresponding absorbance contributions of buffer and free anticancer drug solutions were recorded and digitally subtracted with the same instrumental parameters. All spectra were taken via the ATR method with a resolution of 4 cm−1. The subtraction of the reference spectrum from the spectrum of the protein solution was carried out in accord with the criteria that a straight baseline was obtained between 2000 and 1750 cm−1.28 The concentration of the anticancer drug was equal to HSA in the FT-IR study.2.6 CD spectra measurements
For the CD experiment, the concentration of HSA was kept at 2.0 × 10−6 mol L−1, while the concentration of anticancer drug was changed from 0 to 1.4 × 10−5 mol L−1. The CD spectra were recorded from 200 to 260 nm in 0.1 M PBS (pH 7.4) at room temperature under constant nitrogen flush. The CD profiles were obtained employing a scan speed of 500 nm min−1 and response time of 0.5 s. Each spectrum was the average of three successive scans and was corrected by PBS buffer solution. Appropriate baseline corrections in the CD spectra were made. The contents of secondary conformation of HSA (α-helix, β-strand, turn and unordered) were analyzed from CD spectroscopic data.2.7 Mass spectra measurements
The UPLC was directly interfaced with a Waters XEVO G2 Q TOF mass system equipped with an electrospray ion source operating in either positive or negative ESI mode. Chromatographic separation was achieved on ACQUITY UPLC BEH300 C4 column (1.7 μm, 2.1 × 100 mm) at 80 °C. The mobile phase consisted of a constant flow rate gradient of methanol 0.1% formic acid in water and 0.1% formic acid in acetonitrile under an elution program at a flow rate of 0.2 mL min−1. The analysis run time was 12 min for each injection and the injection volume was 10 μL. The optimal conditions of analysis were as follows: ESI+ mode, capillary voltage of 2.5 kV, sampling cone voltage was 30.0 V, extraction cone voltage was 4.0 V. The temperature was set at 100 °C, desolvation gas temperature was 350 °C and desolvation gas flow was 800 L h−1. The mass spectrometer was calibrated using a solution of sodium formate before the experiment. The full-scan mass spectral data were produced across the mass range of 500–4000 Da. Scans were of 12 min duration. Data were collected in centroid mode and mass was corrected during acquisition using an external reference (Lock-Spray) comprising a 200 pg mL−1 solution of leucine-enkephalin via a lockspray interface, generating a reference ion at 556.277 Da ([M + H]+) for positive ESI mode.For these experiments, the HSA concentration was fixed at 2.0 × 10−4 mol L−1 and the anticancer drug concentration at 3.0 × 10−3 mol L−1. Free HSA and drug–HSA (molar ratio of 1:1 and 2:1) system were prepared in 0.1% formic acid in acetonitrile and introduced into the mass spectrometer source with a syringe pump. The mass spectral data of all determined samples were further processed by BiopharmLynx V1.3.3 software (Waters Corporation, Milford, USA) for peak detection and peak alignment. The method parameters for data processing were set as follows: retention time range 4.55–8 min, deconvolution m/z range 1190–1480 Da, protein molecular mass range 62000–75000 Da, mass tolerance 100 ppm, noise elimination level 6 and peak intensity threshold 50.
2.8 Cyclic voltammetry measurements
The bare gold electrode was successively polished by α-Al2O3 powder with the diameter of 1.0 μm, 0.3 μm and 0.05 μm, respectively. Subsequently, the bare gold electrode was ultrasonically cleaned in acetone, 0.5 M H2SO4 solution and ultrapure water, and then dried by nitrogen airflow. Then, 5 μL HSA solution (2.0 × 10−6 mol L−1) was dropped to the surface of the bare gold electrode. After 6 h, the electrode was washed by ultrapure water several times and dried in nitrogen airflow and was then ready for use. All three of the electrodes were fixed by the O-ring onto a panel constructed from virgin Teflon. The electrolyte used in the experiments was a solution of 1:1 5.0 × 10−3 mol L−1 K3Fe(CN)6/K4Fe(CN)6, and 10 mL of electrolyte was added into an electrochemical cell with a magnetic stirring device. After that, different amounts of the anticancer drug solution were added continuously to the system and stirred for 5 min; then, the system was at rest for 3 min before testing. The cyclic voltammograms of HSA modified gold electrode in the absence and the presence of drug were recorded at a scan rate of 50 mV s−1, with the concentration of anticancer drug varied from 1.2 × 10−7 mol L−1 to 9.6 × 10−7 mol L−1. All measurements were repeated three times with different electrodes at room temperature.
3. Results and discussion
3.1 Fluorescence quenching investigation
It is well-know that the intrinsic fluorescence of HSA is mostly contributed by Try residue alone and is very sensitive to its local microenvironment. The interaction of other molecules and HSA could result in the intrinsic fluorescence quenching of HSA obviously.29 The fluorescence quenching is mainly occurred by collisional process (dynamic quenching) and/or ground state complex formation by quencher and fluorophore (static quenching). Since the steady-state fluorescence spectroscopy is widely used to obtain the local information about both microenvironmental and conformational changes of fluorophore,30,31 which can further confirm the fluorescence quenching mechanism. So, the fluorescence spectra of HSA with different concentrations of [(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl anticancer drug were recorded by the steady-state fluorescence spectroscopy, which was in order to know whether drug formed a complex with HSA or just collided with HSA by accident.The effect of this anticancer drug on the fluorescence intensity of HSA at 298 K was shown in Fig. 1. It could be seen that HSA showed a strong fluorescence emission band around 350 nm in the wavelength range of 300–450 nm and under the excitation wavelength of 295 nm, while the anticancer drug showed almost no fluorescence emission in the same conditions (line L in Fig. 1). Furthermore, the fluorescence intensity of HSA was quenched regularly with the increase of the anticancer drug concentration, and the fluorescence intensity of HSA decreased dramatically when the anticancer drug with higher concentration was present in the solution, which suggested that the anticancer drug could interact with HSA and then quench the intrinsic fluorescence of HSA by concentration-dependent manner.22,23 Same trends could also be observed at other temperatures (304 K and 310 K), but the fluorescence emission positions and the shapes of fluorescence spectra were almost not changed in the whole process.
3.2 Fluorescence quenching mechanism
The possible fluorescence quenching mechanism might be either static or dynamic or both of them that could be distinguished easily by the difference on temperature dependence. The quenching constants decrease with the increment of temperature for static quenching while the reverse is true for dynamic quenching. Since the fluorescence quenching data at different temperature are plotted as the relative fluorescence intensity of HSA versus the anticancer drug concentration, the quenching constants of different temperature can be calculated through the well-known Stern–Volmer equation shown below:32
 | (1) |
In the above equation, F0 and F represent the steady-state fluorescence intensities of fluorophore at the absent and present of quencher, respectively. kq is the bimolecular quenching constant and the maximum value of kq is 2.0 × 1010 L mol−1 s−1 for dynamic quenching, τ0 is the fluorescent lifetime of fluorophore in the absence of quencher and τ0 is approximately 1.0 × 10−8 s for HSA. KSV is the Stern–Volmer quenching constant and [Q] is the concentration of quencher. So, the Stern–Volmer quenching constant KSV value can be calculated by the linear regression of a plot of F0/F against [Q], and kq value can be calculated by the ratio of KSV/τ0 from the above equation.
In order to evaluate the dilution effect by buffer solutions, the measurement of HSA titrated by buffer was performed. It was observed that the fluorescence spectra of HSA were almost not varied during the buffer titration process. The plots of Stern–Volmer equation at different temperature were shown in Fig. 2A, and the values of Stern–Volmer quenching constants KSV and bimolecular quenching constants kq at three different temperatures were listed in Table 1. As shown in Fig. 2A, the results agreed well with the Stern–Volmer equation at lower concentrations (0–1.0 × 10−6 mol L−1 with an interval of 1.0 × 10−7 mol L−1), since the results could depart from the initial linearity at higher concentrations due to the inner filter effects.33 Furthermore, as indicated in Table 1 that both the KSV and kq values increased with the temperature rising, indicating that the probable quenching mechanism of HSA by this anticancer drug could be a dynamic quenching rather than static quenching. However, the kq values were larger than 2.0 × 1010 L mol−1 s−1 all (the maximum value for dynamic quenching), suggesting that the quenching mechanism might be a static quenching mechanism.
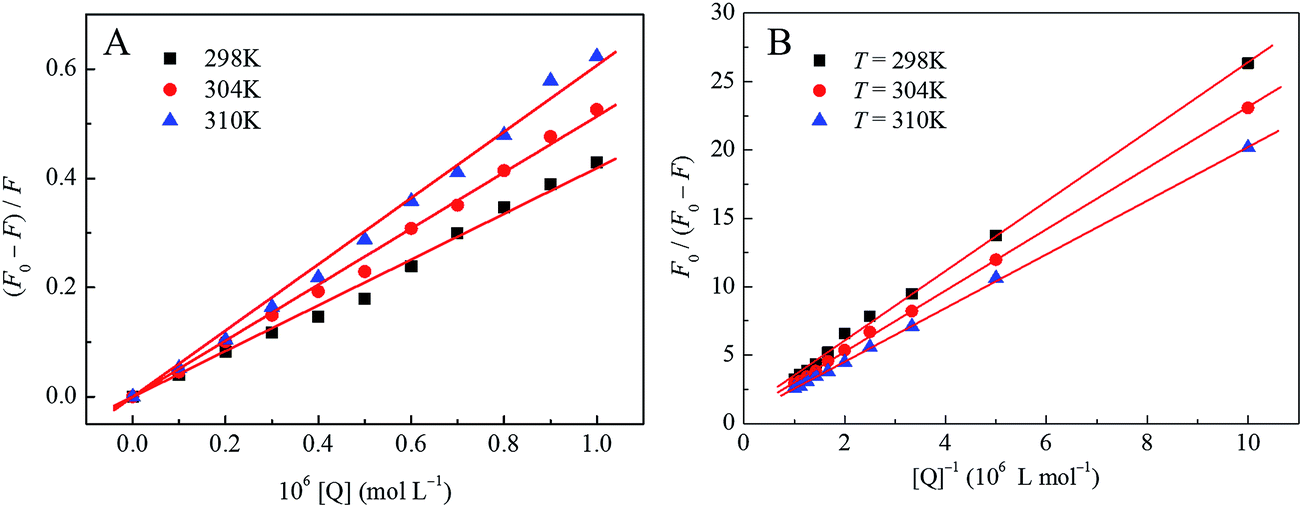 | ||
| Fig. 2 The Stern–Volmer plots of drug–HSA system (A) and the modified Stern–Volmer plots of drug–HSA system (B) at three different temperatures. | ||
| pH | T/(K) | KSV (105 L mol−1) | Kq (1013 L mol−1 s−1) | R2a | S.D.b | Ka (105 L mol−1) | R2a | ΔH (kJ mol−1) | ΔG (kJ mol−1) | ΔS (J mol−1 K−1) | R2a | S.D.b |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a R2 is the correlation coefficient.b S.D. is standard deviation. | ||||||||||||
| 7.4 | 298 | 4.19 | 4.19 | 0.996 | 0.014 | 3.67 | 0.998 | −10.75 | −31.74 | 70.43 | 0.999 | 0.012 |
| 304 | 5.14 | 5.14 | 0.998 | 0.012 | 3.38 | 0.999 | −32.17 | |||||
| 310 | 6.07 | 6.07 | 0.997 | 0.017 | 3.10 | 0.999 | −32.58 | |||||
It is well known that the dynamic quenching can only affect the excited states of fluorophores and no obvious variations in the absorption spectra of fluorophores are expected, so the UV-vis absorption spectra are further performed to identify the real quenching mechanism of this procedure.34 As shown in Fig. 3, the UV-vis absorption spectrum of HSA (curve B in Fig. 3) in the near ultraviolet band (200–350 nm) and the difference absorption spectrum between drug–HSA system and anticancer drug alone (curve D in Fig. 3) at the same range could not be superposed within experimental error, indicating the ground state complex formation between this anticancer drug and HSA rather than the dynamic collision. The results could further confirm that the fluorescence quenching mechanism of HSA by this anticancer drug was mainly a static quenching mechanism.
 | ||
| Fig. 3 UV-visible spectra of HSA in the presence of this anticancer drug. (A) The absorption spectrum of this anticancer drug only; (B) the absorption spectrum of HSA only; (C) the absorption spectrum of drug–HSA system when the mole ratio is 1:1; (D) the difference absorption spectrum between drug–HSA system and drug at the same concentration. c (HSA) = c (drug) = 2.0 × 10−6 mol L−1. | ||
For the static quenching process, the quenching data could be further analyzed according to the modified Stern–Volmer equation:32
 | (2) |
In the present case, F0 is the fluorescence intensity of fluorophore in the absence of quencher, and ΔF is the difference of the fluorescence intensities of fluorophore in the absence and presence of quencher. fa is the mole fraction of solvent accessible fluorophore, Ka is the effective quenching constant for the accessible fluorophores and [Q] is the concentration of quencher,35 respectively, which are analogous to associative binding constants for the quencher–acceptor system.36,37 The dependence of F0/ΔF on the reciprocal value of quencher concentration [Q]−1 is linear with the slope equaling the value of (faKa)−1, and the value of fa−1 is fixed on the ordinate. The modified Stern–Volmer quenching constant Ka can be calculated from the quotient of the ordinate fa−1 and the slope (faKa)−1. The plots of modified Stern–Volmer equation were shown in Fig. 2B, and the corresponding modified Stern–Volmer quenching constants Ka at three different temperatures were also listed in Table 1. The decreasing trend of Ka with increasing temperature indicated that the fluorescence quenching was mainly arisen from static quenching by complex formation.32
3.3 The investigation of the interaction between drug and HSA
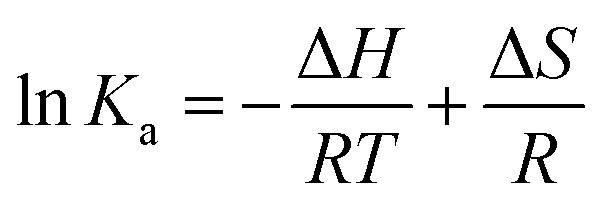 | (3) |
Ka and 1/T was existed. According to the slopes and the intercepts as shown in Fig. 4, the values of ΔH and ΔS could be calculated and the results were also shown in Table 1. Furthermore, the free energy change (ΔG) can be calculated from the below equation:| ΔG = ΔH − TΔS | (4) |
 | ||
| Fig. 4 van't Hoff plots of drug–HSA system. | ||
The values of ΔG at different temperatures were all incorporated in Table 1. The negative values of ΔG and the positive value of ΔS indicated that the binding process of drug to HSA was spontaneous. The negative ΔH and the positive ΔS values of the interactions of drug and HSA suggested that the electrostatic interactions played major roles in this binding reaction,39 which was agreed well with the conclusion reported before.40 Moreover, hydrophobic interactions and coordination binding might also participate in the interaction of HSA with this anticancer drug.
 | (5) |
In the present case, F0 and F are the fluorescence intensities of fluorophore in the absence and the presence of quencher, respectively. Kb is the binding constant and n is the binding number of the fluorophore and quencher system, which can be abstained by the plot of log(F0 − F)/F versus log[Q]. Table 2 demonstrates the results analyzed by this way for drug–HSA system at different temperatures. It could be seen that the value of Kb increased dramatically with the increase of temperatures, while n was almost kept constant, indicating that this anticancer drug bound strongly with HSA according to mole ratio 1:1, and independent of the binding site for this anticancer drug to HSA.
| Systems | Kb (105 L mol−1) | n | Ra | S.D.b | K/K0c |
|---|---|---|---|---|---|
| a R2 is the correlation coefficient.b S.D. is standard deviation.c K0 and K are the apparent binding constants of drug–HSA system alone and drug–HSA system in the absence of metal ion, respectively. | |||||
| Drug–HSA | 7.16 | 1.09 | 0.996 | 0.028 | 1 |
| Drug–HSA–Ag+ | 1.76 | 0.96 | 0.998 | 0.019 | 0.247 |
| Drug–HSA–K+ | 5.99 | 1.05 | 0.999 | 0.012 | 0.837 |
| Drug–HSA–Na+ | 7.67 | 1.07 | 0.999 | 0.014 | 1.071 |
| Drug–HSA–Cu2+ | 2.93 | 0.99 | 0.999 | 0.012 | 0.409 |
| Drug–HSA–Zn2+ | 6.27 | 1.06 | 0.999 | 0.015 | 0.875 |
| Drug–HSA–Ca2+ | 3.80 | 1.02 | 0.998 | 0.018 | 0.532 |
| Drug–HSA–Mg2+ | 8.57 | 1.07 | 0.997 | 0.012 | 1.197 |
| Drug–HSA–Ni2+ | 1.07 | 0.93 | 0.999 | 0.027 | 0.150 |
| Drug–HSA–Co2+ | 6.47 | 1.06 | 0.999 | 0.013 | 0.904 |
| Drug–HSA–NO3− | 1.76 | 0.97 | 0.998 | 0.019 | 0.247 |
| Drug–HSA–SO42– | 2.93 | 0.99 | 0.999 | 0.012 | 0.409 |
| Drug–HSA–Cl− | 1.07 | 0.93 | 0.999 | 0.027 | 0.150 |
 | ||
| Fig. 5 Effect of site-specific probe to drug–HSA system, (A) warfarin; (B) ibuprofen (pH 7.4, T = 298 K, λex = 295 nm). (C) The modified Stern–Volmer plots of drug–HSA system alone and with warfarin or ibuprofen probe. c (warfarin) = c (ibuprofen) = c (HSA) = 2.0 × 10−6 mol L−1; c (drug)/(10−6 mol L−1), 1–11: 0; 0.1; 0.2; 0.3; 0.4; 0.5; 0.6; 0.7; 0.8; 0.9; 1.0; the inserts correspond to the molecular structures of site-specific probe. | ||
In order to facilitate the comparison of the influence of site-specific probes on the fluorescence quenching of HSA by this anticancer drug, the Stern–Volmer quenching constants of drug–HSA–probe system were further analyzed using the eqn (1). The Stern–Volmer quenching constant values at 298 K were noticed to be 4.19 × 105 L mol−1, 1.83 × 105 L mol−1 and 3.72 × 105 L mol−1 for drug–HSA system, drug–HSA–warfarin system and drug–HSA–ibuprofen system, respectively. As evident from the results, the Stern–Volmer quenching constant was all decreased at the present of warfarin or ibuprofen but it was decreased dramatically at the present of warfarin than ibuprofen, which indicted that this anticancer drug could be significantly displaced by warfarin and the binding site of this anticancer drug was mainly located within site I. Hence, the site I, which has been described as a large hydrophobic cavity present in subdomain IIA of HSA and involved the lone Trp214 residue of HSA,45 was proposed to be the main binding site for this anticancer drug in HSA. Furthermore, it is reported before that HSA has negative net charge of about −15, while it contains three subdomains with different net charges under the physiological pH: −9 (subdomain I), −8 (subdomain II), and +2 (subdomain III).46 This anticancer drug in solution was positively charged and could bind to the surface of subdomain I and subdomain II spontaneously, which demonstrated that the electrostatic interaction was the main force in the interaction between HSA and this anticancer drug.
3.4 Energy transfer between HSA and drug
Since HSA contains the lone Trp214 in subdomain IIA, the distance between the binding site of this anticancer drug and the fluorophore (Trp214) of HSA could be calculated according to Förster theory of fluorescence resonance energy transfer (FRET). According to the Förster theory of FRET, both the efficiency of energy transfer (E) and the distance (r) between the donor (Trp214 in HSA) and the acceptor (drug) could be calculated by the equation shown below:32
 | (6) |
| R06 = 8.79 × 10−25 K2n−4ϕJ | (7) |
In the above equation, K2 is the space factor of orientation related to the geometry of the donor and the acceptor of dipoles and K2 = 2/3 for random orientation in solution; n is the refracted index of medium and n = 1.36 usually; ϕ is the fluorescence quantum yield of the donor without the acceptor and ϕ = 0.074 for HSA;47 J is the degree of spectral overlap between the emission spectrum of the donor and the absorption spectrum of the acceptor (Fig. 6), which can be calculated by the equation:
 | (8) |
 | ||
| Fig. 6 Overlapping between the UV-vis absorption spectrum of this anticancer drug (a) and the fluorescence emission spectrum of HSA (b) (λex = 295 nm). c (HSA) = c (drug) = 2.0 × 10−6 mol L−1. | ||
Herein, F(λ) is the corrected fluorescence intensity of the donor in the wavelength range of λ to λ + Δλ; ε(λ) is the extinction coefficient of the acceptor at λ.
According to above equations, the calculated values of R0 and r were 4.31 nm and 3.68 nm, respectively. The absolute value of the average distance between the donor and the acceptor was in the range of 2–8 nm and 0.5R0 < r < 1.5R0,48 indicating the higher probability of occurrence of the energy transfer from HSA to this anticancer drug, which was also in accordance with the electrostatic interaction mechanism.
3.5 Conformation change investigation
The influence of this anticancer drug on the synchronous fluorescence spectrum of HSA applied to the microenvironment investigation of Tyr residue and Trp residue was shown in Fig. 7. It was apparent from Fig. 7 that the maximum emission wavelength of Tyr residue (Δλ = 15 nm) showed a subtle red shift (from 299 nm to 302 nm), while the maximum emission wavelength of Trp residue (Δλ = 60 nm) exhibited a tiny blue shift (from 340 nm to 339 nm). These results suggested that the binding of this anticancer drug to HSA could affect the microenvironment around Tyr and Trp,50 and the Tyr residue became much more hydrophobic while the Trp residue became much more hydrophilic. As shown in Fig. 7C, the curve of Δλ = 60 nm was lower than that of Δλ = 15 nm, indicating that Trp residue played an important role during the fluorescence quenching of HSA by this anticancer drug and the anticancer drug could approach the Trp residue more than the Tyr residue.51
 | ||
| Fig. 7 Synchronous fluorescence spectrum of drug–HSA system, (A) Δλ = 15 nm; (B) Δλ = 60 nm. (C) Quenching of HSA synchronous fluorescence by this anticancer drug. c (HSA) = 2.0 × 10−6 mol L−1; c (drug)/(10−6 mol L−1); 1–13: 0; 0.4; 0.8; 1.2; 1.6; 2.0; 2.4; 2.8; 3.2; 3.6; 4.0; 4.4; 4.8. | ||
 | ||
| Fig. 8 Three-dimensional fluorescence spectra of HSA alone (A) and drug–HSA system (B). | ||
| Peaks | HSA | Drug–HSA system | ||||
|---|---|---|---|---|---|---|
| Peak position λex/em (nm nm−1) | Stokes shift Δλ (nm) | Intensity F | Peak position λex/em (nm nm−1) | Stokes shift Δλ (nm) | Intensity F | |
| Fluorescence peak 1 | 285.0/346.6 | 61.6 | 627.5 | 285.0/346.1 | 61.1 | 438.2 |
| Fluorescence peak 2 | 230.0/345.9 | 115.9 | 614.4 | 245.0/349.4 | 104.4 | 158.3 |
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch) and amide II band (1500–1550 cm−1, C–N stretch coupled with N–H bending mode) both related with the secondary structure of proteins.22 Compared with the amide II band, the amide I band is more sensitive to the variation of the secondary structure of proteins. The FT-IR spectra of free HSA and drug–HSA system in PBS were shown in Fig. 9. As could be seen from infrared spectrum of free HSA, the peak positions of the amide I band and the amide II band were observed at 1652.5 cm−1 and 1548.2 cm−1, respectively. When this anticancer drug was present in HSA solution, the peak position of amide I band moved from 1652.5 cm−1 to 1658.1 cm−1 and the peak position of amide II band moved from 1548.2 cm−1 to 1544.0 cm−1, together with the changes in the peak intensities of both amide I band and amide II band. These results indicated that this anticancer drug could interact with CO, C–N and N–H groups in the protein polypeptides that resulted in the rearrangement of the secondary structure of HSA.52
O stretch) and amide II band (1500–1550 cm−1, C–N stretch coupled with N–H bending mode) both related with the secondary structure of proteins.22 Compared with the amide II band, the amide I band is more sensitive to the variation of the secondary structure of proteins. The FT-IR spectra of free HSA and drug–HSA system in PBS were shown in Fig. 9. As could be seen from infrared spectrum of free HSA, the peak positions of the amide I band and the amide II band were observed at 1652.5 cm−1 and 1548.2 cm−1, respectively. When this anticancer drug was present in HSA solution, the peak position of amide I band moved from 1652.5 cm−1 to 1658.1 cm−1 and the peak position of amide II band moved from 1548.2 cm−1 to 1544.0 cm−1, together with the changes in the peak intensities of both amide I band and amide II band. These results indicated that this anticancer drug could interact with CO, C–N and N–H groups in the protein polypeptides that resulted in the rearrangement of the secondary structure of HSA.52
 | ||
| Fig. 9 ATR FT-IR spectra of HSA and drug–HSA system in PBS of pH 7.4 solution: (A) FT-IR spectrum of HSA only; (B) FT-IR spectra of drug–HSA system. All the spectra spanned the 1800–1450 cm−1 regions. | ||
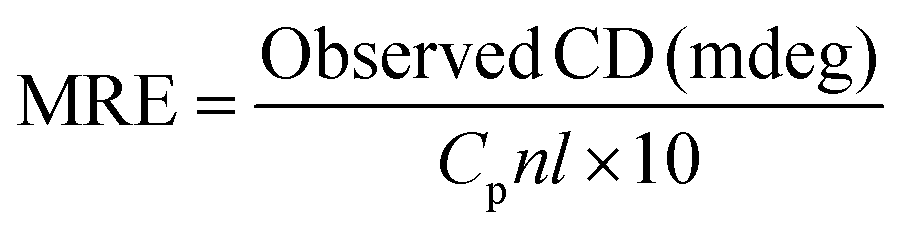 | (9) |
 | (10) |
Herein, MRE208 is the observed MRE value at 208 nm. 4000 represents the MRE value of the β-form and random coil conformation cross at 208 nm and 33000 means the MRE value of a pure α-helix at 208 nm, respectively. From the above equation, the α-helicity in the secondary structure of HSA could be easily determined.
As shown in Fig. 10, HSA exhibited two negative absorption bands in the far-UV region at 208 nm and 222 nm, which was consistent with the results reported before. However, the CD intensity at 208 nm was decreased dramatically and the shape of the CD spectrum was also changed significantly after the addition of the anticancer drug. In order to quantify the different contents of the secondary structures of HSA, the CD spectra were analyzed by the algorithm SELCON3 and the values of secondary structures for HSA were also calculated. As shown in Table 5, a decreasing tendency of the α-helices content and increasing tendencies of other secondary structure contents were estimated at higher concentration of this anticancer drug. The α-helices content of HSA decreased from 60.1% to 43.4%, while the β-strand content of HSA increased from 6.0% to 8.2% and the unordered content of HSA also increased from 20.1% to 24.8%, when the concentration of this anticancer drug increased from 0 to 1.4 × 10−5 mol L−1, which indicated that the decrease of the biological activity of HSA upon the interaction with this anticancer drug of higher concentration. In addition, the decrease of the α-helices content in the secondary structure of HSA also indicated that this anticancer drug could enter the hydrophobic pocket of HSA and destroyed the hydrogen bonding networks of HSA. These results further elucidated that a stronger structural change of that was related to a low degree of surface coverage.54
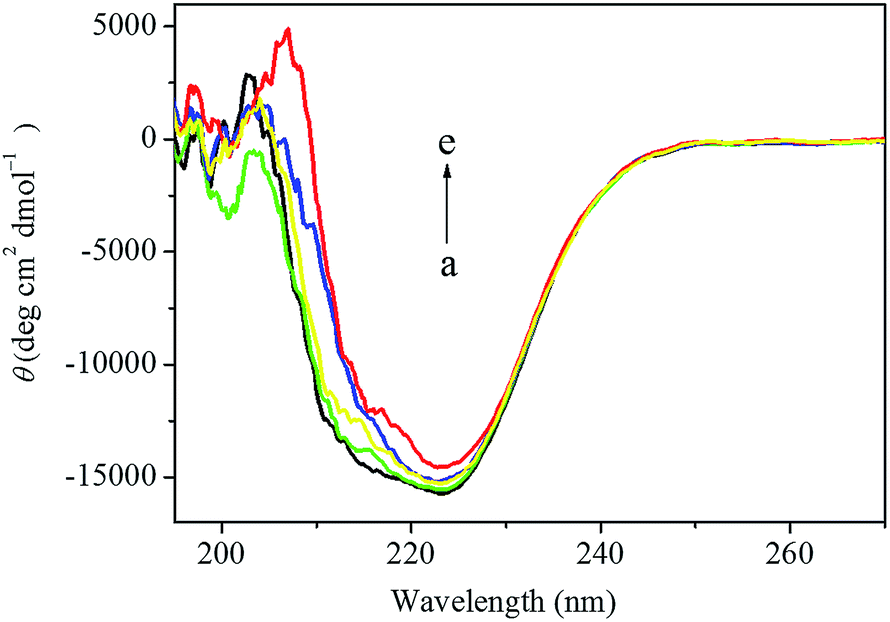 | ||
| Fig. 10 The CD spectra of the drug–HSA system obtained in 0.1 mol L−1 PBS of pH 7.4 at room temperature. The concentration of HSA was fixed at 2.0 × 10−6 mol L−1 (a). In drug–HSA system, the concentrations of this anticancer drug were 2.0 × 10−6 mol L−1 (b), 6.0 × 10−6 mol L−1 (c), 1.0 × 10−5 mol L−1 (d) and 1.4 × 10−5 mol L−1 (e), respectively. | ||
| Concentration of the anticancer drug (mol L−1) | Percentage of secondary structure | |||||
|---|---|---|---|---|---|---|
| α-helix | β-strand | Turn | Unordered | |||
| αR | αD | βR | βD | |||
| a The subscripts “R” and “D” represent “ordered” and “disordered”, respectively. | ||||||
| 0 | 42.2 | 17.9 | 3.1 | 2.9 | 13.8 | 20.1 |
| 2.0 × 10−6 | 38.8 | 17.2 | 3.5 | 3.2 | 15.2 | 22.1 |
| 6.0 × 10−6 | 35.7 | 16.6 | 3.8 | 3.4 | 18.1 | 22.4 |
| 1.0 × 10−5 | 31.2 | 15.4 | 4.0 | 3.8 | 22.1 | 23.5 |
| 1.4 × 10−5 | 28.5 | 14.9 | 4.2 | 4.0 | 23.6 | 24.8 |
3.6 Mass spectra investigation
The ground state complexation of exogenous ligands with HSA could be further demonstrated by using XEVO G2 Q TOF mass spectrometry which was considered to be an accurate and sensitive approach for the verification of the binding interaction between exogenous ligands and HSA.55 In order to prove the static quenching mechanism between drug and HSA, the mass spectra of free anticancer drug, free HSA and drug–HSA complexes were performed and all depicted in Fig. 11. It was easily to know that the precise molecular mass of anticancer drug was 561.53 according to the molecular formula, but in the positive ion spectrum, the main fragment ion of anticancer drug was observed at m/z 490 (Fig. 11A) that was corresponded with the molecular mass of the structure of [M − Cl − HCl]+.11 | ||
| Fig. 11 The mass spectra of anticancer drug alone (A), HSA alone (B), the drug–HSA system with molar ratio of 1:1 (C) and 2:1 (D), respectively. | ||
According to Fig. 11B, it could be seen that the average molecular mass of HSA alone was around 66438 Da, which was in accordance with the reported value.55 The same molecular mass of 66438 Da could also be seen when the anticancer drug was present (Fig. 11C). Furthermore, after the addition of anticancer drug, new peaks corresponded with different molecular mass occurred at about 66927 Da and 66962 Da, and the molecular mass was increased by 491 Da and 524 Da compared with the molecular mass of HSA alone. Since the molecular weight of the main fragment ion of anticancer drug in the positive ion spectrum was 490 Da, indicating that this anticancer drug was indeed binding to HSA. In addition, the anticancer drug could easily detach a chloride ion and existed in the structure of [M − Cl]+ (molecular mass 526 Da) in solution,56 so the positively-charged anticancer drug could easily bind with the negatively-charged HSA in physiological conditions and then the molecular mass of around 66964 Da appeared spontaneously. These results were a clear indication that the additional molecular mass on HSA originated from one molecule of anticancer drug ligand. Moreover, the same molecular mass could be seen in drug–HSA system with different molecular ratio of anticancer drug versus HSA (Fig. 11C and D), which further revealed that the binding number of anticancer drug to HSA was 1:1 that was consistent with the fluorescent spectrometry result. Thus, this additional molecular mass of one anticancer drug molecule revealed the presence of a single anticancer drug molecule associated with HSA on average.
3.7 Cyclic voltammetry investigation
Cyclic voltammetry is an effective method to directly analyze the interaction between proteins and drugs under the physiological conditions.16,57 In order to further confirm the results obtained from the spectroscopic methods, cyclic voltammetry is used to investigate the interaction between this anticancer drug and HSA at gold electrode. Since HSA need to be fixed on the surface of gold electrode, the HSA-modified gold electrode is characterized firstly by using K3Fe(CN)6/K4Fe(CN)6 probe which shows different electrochemical behavior on different films. As indicated in Fig. 12, a good electrochemical response of K3Fe(CN)6/K4Fe(CN)6 with a pair of reversible redox peaks existed on both bare gold electrode and HSA-modified gold electrode. However, the formal redox peak potentials negatively shifted and the formal redox peak currents dramatically decreased, showing the great variation occurred on the surface of the gold electrode. Furthermore, after the modification of HSA on the surface of gold electrode, the separation of redox peak potential increased from 84 mV to 97 mV and the separation of redox peak current decreased from 83.4 μA to 80.7 μA, respectively, indicating the decrease of both the electron-transfer rate of K3Fe(CN)6/K4Fe(CN)6 and the conductivity of the modified gold electrode. In addition, the gold electrode treated with HSA could be reused with the constant redox peak potentials and redox peak currents, demonstrating the immobilization of HSA on the surface of gold electrode. | ||
| Fig. 12 Cyclic voltammograms of bare Au electrode and HSA-modified Au electrode in the absence and the presence of various concentrations of this anticancer drug. c (drug)/(10−7 mol L−1); 1–9: 0, 1.2, 2.4, 3.6, 4.8, 6.0, 7.2, 8.4, 9.6. Insert was the linear relationship existed between the reciprocal of the current drop and the reciprocal of this anticancer drug concentration in this range. | ||
This HSA-modified gold electrode was subsequently used as the working electrode to investigate the interaction between this anticancer drug and HSA. As shown in Fig. 12, when the anticancer drug was administrated into the K3Fe(CN)6/K4Fe(CN)6 electrolyte solution, a significant decrease in the redox peak currents and a tiny shift in the redox peak potentials were happened. The decrease of the redox peak currents and the shift of the redox peak potentials after the addition of this anticancer drug might result from the interaction between HSA and this anticancer drug that reduced the diffusion coefficient of K3Fe(CN)6/K4Fe(CN)6 subsequently. Furthermore, the lower the redox peak current was generated when the higher the concentration of this anticancer drug was present, which suggested that this anticancer drug could decrease the redox peak currents by concentration-dependent manner. According to values of the redox peak currents in the presence of different concentrations of anticancer drug obtained from Fig. 12, a good linear relationship existed between the reciprocal of the current drop and the reciprocal of the anticancer drug concentration, with a correlation coefficient of 0.998. It was compliance with the Langmuir equation:57
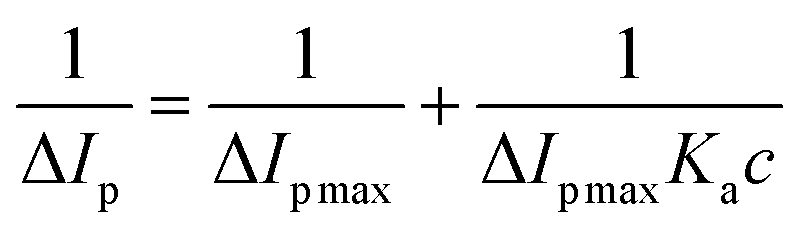 | (11) |
Herein, ΔIp is the current drop, ΔIp max is the maximum of the current drop, Ka is the binding equilibrium constant between HSA and the anticancer drug, and c is the concentration of the anticancer drug, respectively. According to the equation and the slope of the curve insert in Fig. 12, the value of the equilibrium constant Ka was 1.267 × 105 L mol−1, which was a little lower than the value of the binding constant obtained by spectroscopic methods. Moreover, the electrode was washed with ultrapure water and then dipped into fresh K3Fe(CN)6/K4Fe(CN)6 solution, the redox peak currents changed greatly, suggesting the complex formation between this anticancer drug and HSA on the surface of gold electrode, which was consistent well with the results proved by multispectroscopic approaches.
4. Conclusions
During this work, we systematically investigated the interactions between [(η6-p-cymene)Ru(benzaldehyde-N(4)-phenylthiosemicarbazone)Cl]Cl anticancer drug and HSA by UV-vis absorption spectra, fluorescence spectra, FT-IR spectra, CD spectra, mass spectra and cyclic voltammetry under the physiological conditions. The Stern–Volmer quenching constants, the bimolecular quenching constants, the modified Stern–Volmer quenching constants and the binding constants were all obtained. The results indicated that this anticancer drug quenched the intrinsic fluorescence of HSA effectively by concentration-dependent manner, the quenching mainly belonged to the static quenching. The subsequent results indicated that the binding interaction between this anticancer drug and HSA could be affected efficiently by common metal ions. The values of some thermodynamic parameters elucidated that the electrostatic interactions played major roles during their interaction, and this anticancer drug could effectively occupy site I. The synchronous fluorescence spectroscopy, three-dimensional fluorescence spectroscopy, FT-IR spectroscopy and CD spectra were further used to indicate the microenvironmental and conformational changes of HSA with this anticancer drug, which indicated that the microenvironment and the conformation of HSA were changed drastically at the present of this drug and the biological activity of HSA was weakened in the present of this anticancer drug. The results of mass spectra further demonstrated that the binding number of anticancer drug to HSA was 1:1. Cyclic voltammetry further confirmed the interaction between HSA and this anticancer drug. These results made a better understanding on the interactions between ruthenium(II) arene complexes of TSC anticancer drug and serum albumin, which is much important for the further applications of this kind of anticancer drug in biomedical and therapeutical fields.
Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (21203035, 21261005, 51263002), the Guangxi Natural Science Foundation (2013GXNSFCA019005, 2013GXNSFBA019029, 2014GXNSFBA118243), the Scientific Research Foundation of Guangxi Provincial Education Department (2013YB138, ZD2014081), the Scientific Research Foundation for the Returned Overseas Chinese Scholars, State Education Ministry and Open Research Fund Program of the State Key Laboratory of Virology of China (2014KF006).References
- M. Adams, Y. Q. Li, H. Khot, C. D. Kock, P. J. Smith, K. Land, K. Chibale and G. S. Smith, The synthesis and antiparasitic activity of aryl- and ferrocenyl-derived thiosemicarbazone ruthenium(II)-arene complexes, Dalton Trans., 2013, 42, 4677–4685 RSC.
- A. K. Singh, D. S. Pandey, Q. Xu and P. Braunstein, Recent advances in supramolecular and biological aspects of arene ruthenium(II) complexes, Coord. Chem. Rev., 2014, 270–271, 31–56 CrossRef CAS PubMed.
- Q. Zhou, P. Y. Li, R. M. Lu, Q. Q. Qian, X. L. Lei, Q. Xiao, S. Huang, L. F. Liu, C. S. Huang and W. Su, Synthesis, X-ray diffraction study and cytotoxicity of a cationic p-cymene ruthenium chloro complex containing a chelating semicarbazone ligand, Z. Anorg. Allg. Chem., 2013, 639, 943–946 CrossRef CAS PubMed.
- L. F. Liu, P. Y. Li, Q. Q. Qian, X. L. Lei, Y. X. Huang, Q. Zhou, S. Huang, Q. Xiao and W. Su, Synthesis, structure, anticancer activity of 2-formylthiophene thiosemicarbazones and their organometallic ruthenium complexes, Chin. J. Org. Chem., 2013, 33, 854–859 CrossRef CAS.
- F. A. Beckford, G. Leblanc, J. Thessing, M. Jr Shaloski, B. J. Frost, L. Li and N. P. Seeram, Organometallic ruthenium complexes with thiosemicarbazone ligands: synthesis, structure and cytotoxicity of [(η6-p-cymene)Ru(NS)Cl]+ (NS = 9-anthraldehyde thiosemicarbazones), Inorg. Chem. Commun., 2009, 12, 1094–1098 CrossRef CAS PubMed.
- F. A. Beckford, D. Dourth, M. Shaloski Jr, J. Didion, J. Thessing, J. Woods, V. Crowell, N. Gerasimchuk, A. Gonzalez-Sarrías and N. P. Seeram, Half-sandwich ruthenium-arene complexes with thiosemicarbazones: synthesis and biological evaluation of [(η6-p-cymene)Ru(piperonal thiosemicarbazones)Cl]Cl complexes, J. Inorg. Biochem., 2011, 105, 1019–1029 CrossRef CAS PubMed.
- T. Stringer, B. Therrien, D. T. Hendricks, H. Guzgay and G. S. Smith, Mono- and dinuclear (η6-arene) ruthenium(II) benzaldehyde thiosemicarbazone complexes: synthesis, characterization and cytotoxicity, Inorg. Chem. Commun., 2011, 14, 956–960 CrossRef CAS PubMed.
- M. Adams, Y. Li, H. Khot, C. De Kock, P. J. Smith, K. Land, K. Chibalea and G. S. Smith, The synthesis and antiparasitic activity of aryl- and ferrocenyl-derived thiosemicarbazone ruthenium(II)-arene complexes, Dalton Trans., 2013, 42, 4677–4685 RSC.
- B. Demoro, C. Sarniguet, R. Sanchez-Delgado, M. Rossi, D. Liebowitz, F. Caruso, C. Olea-Azar, V. Moreno, A. Medeiros, M. A. Comini, L. Otero and D. Gambino, New organoruthenium complexes with bioactive thiosemicarbazones as co-ligands: potential anti-trypanosomal agents, Dalton Trans., 2012, 41, 1534–1543 RSC.
- B. Demoro, R. F. M. de Almeida, F. Marques, C. P. Matos, L. Otero, J. C. Pessoa, I. Santos, A. Rodríguez, V. Moreno, J. Lorenzo, D. Gambino and A. I. Tomaz, Screening organometallic binuclear thiosemicarbazone ruthenium complexes as potential anti-tumour agents: cytotoxic activity and human serum albumin binding mechanism, Dalton Trans., 2013, 42, 7131–7146 RSC.
- W. Su, Q. Zhou, Y. M. Huang, Q. Y. Huang, L. N. Luo, Q. Xiao, S. Huang, C. S. Huang, R. Chen, Q. Q. Qian, L. F. Liu and P. Y. Li, Synthesis, crystal and electronic structure, anticancer activity of ruthenium(II) arene complexes with thiosemicarbazones, Appl. Organomet. Chem., 2013, 27, 307–312 CrossRef CAS PubMed.
- W. Su, Q. Q. Qian, P. Y. Li, X. L. Lei, Q. Xiao, S. Huang, C. S. Huang and J. G. Cui, Synthesis, characterization, and anticancer activity of a series of ketone-N4-substituted thiosemicarbazones and their ruthenium(II) arene complexes, Inorg. Chem., 2013, 52, 12440–12449 CrossRef CAS PubMed.
- P. Hanczyc, P. Lincoln and B. Norden, Interactions of binuclear ruthenium(II) complexes with oligonucleotides in hydrogel matrix: enantioselective threading intercalation into GC context, J. Phys. Chem. B, 2013, 117, 2947–2954 CrossRef CAS PubMed.
- A. Kurzwernhart, W. Kandioller, E. A. Enyedy, M. Novak, M. A. Jakupec, B. K. Keppler and C. G. Hartinger, 3-Hydroxyflavones vs. 3-hydroxyquinolinones: structure–activity relationships and stability studies on Ru(II) (arene) anticancer complexes with biologically active ligands, Dalton Trans., 2013, 42, 6193–6202 RSC.
- P. Anitha, N. Chitrapriya, Y. J. Jang and P. Viswanathamurthi, Synthesis, characterization, DNA interaction, antioxidant and anticancer activity of new ruthenium(II) complexes of thiosemicarbazone/semicarbazone bearing 9,10-phenanthrenequinone, J. Photochem. Photobiol., B, 2013, 129, 17–26 CrossRef CAS PubMed.
- J. Wang, C. Xiang, F. F. Tian, Z. Q. Xu, F. L. Jiang and Y. Liu, Investigating the interactions of a novel anticancer delocalized lipophilic cation and its precursor compound with human serum albumin, RSC Adv., 2014, 4, 18205–18216 RSC.
- Y. Zhang, K. Pan and Q. X. Zhong, Characteristics of activated carbon and carbon nanotubes as adsorbents to remove annatto (norbixin) in cheese whey, J. Agric. Food Chem., 2013, 61, 9230–9240 CrossRef CAS PubMed.
- Y. Zhang and Q. X. Zhong, Probing the binding between norbixin and dairy proteins by spectroscopy methods, Food Chem., 2013, 139, 611–616 CrossRef CAS PubMed.
- X. J. Li, B. L. Zhang, W. Li, X. F. Lei, X. L. Fan, L. Tian, H. P. Zhang and Q. Y. Zhang, Preparation and characterization of bovine serum albumin surface-imprinted thermosensitive magnetic polymer microsphere and its application for protein recognition, Biosens. Bioelectron., 2014, 51, 261–267 CrossRef CAS PubMed.
- Q. Xiao, S. Huang, Y. Liu, F. F. Tian and J. C. Zhu, Thermodynamics, conformation and active sites of the binding of Zn–Nd hetero-bimetallic Schiff base to bovine serum albumin, J. Fluoresc., 2009, 19, 317–326 CrossRef CAS PubMed.
- O. K. Abou-Zied, Spectroscopy of hydroxyphenyl benzazoles in solution and human serum albumin: detecting flexibility, specificity and high affinity of the warfarin drug binding site, RSC Adv., 2013, 3, 8747–8755 RSC.
- Q. Xiao, S. Huang, Z. D. Qi, B. Zhou, Z. K. He and Y. Liu, Conformation, thermodynamics and stoichiometry of HSA adsorbed to colloidal CdSe/ZnS quantum dots, Biochim. Biophys. Acta, Proteins Proteomics, 2008, 1784, 1020–1027 CrossRef CAS PubMed.
- Q. Xiao, S. Huang, W. Su, P. Y. Li, J. Q. Ma, F. P. Luo, J. Chen and Y. Liu, Systematically investigations of conformation and thermodynamics of HSA adsorbed to different sizes of CdTe quantum dots, Colloids Surf., B, 2013, 102, 76–82 CrossRef CAS PubMed.
- D. W. Li, H. He, B. B. Lin, Z. Q. Xu, F. L. Jiang and Y. Liu, Studies on the isolated mitochondrial damage induced by α-tocopheryl succinate and its interactions with human serum albumin, RSC Adv., 2013, 4, 3913–3919 RSC.
- Y. P. Zhang, M. K. Nie, S. Y. Shi, Q. P. You, J. F. Guo and L. L. Liu, Integration of magnetic solid phase fishing and off-line two-dimensional high-performance liquid chromatography–diode array detector–mass spectrometry for screening and identification of human serum albumin binders from Radix Astragali, Food Chem., 2014, 146, 56–64 CrossRef CAS PubMed.
- X. R. Li, D. J. Chen, G. K. Wang and Y. Lu, Study of interaction between human serum albumin and three antioxidants: ascorbic acid, α-tocopherol, and proanthocyanidins, Eur. J. Med. Chem., 2013, 70, 22–36 CrossRef CAS PubMed.
- G. J. Zhang, B. Keita, C. T. Craescu, S. Miron, P. D. Oliveira and L. Nadjo, Polyoxometalate binding to human serum albumin: a thermodynamic and spectroscopic approach, J. Phys. Chem. B, 2007, 111, 11253–11259 CrossRef CAS PubMed.
- A. C. Dong, P. Huang and W. S. Caughey, Protein secondary structures in water from second-derivative amide I infrared spectra, Biochemistry, 1990, 29, 3303–3308 CrossRef CAS.
- B. T. Yin, C. Y. Yan, X. M. Peng, S. L. Zhang, S. Rasheed, R. X. Geng and C. H. Zhou, Synthesis and biological evaluation of a-triazolyl chalcones as a new type of potential antimicrobial agents and their interaction with calf thymus DNA and human serum albumin, Eur. J. Med. Chem., 2014, 71, 148–159 CrossRef CAS PubMed.
- Q. Xiao, H. N. Qiu, S. Huang, C. S. Huang, W. Su, B. Q. Hu and Y. Liu, Systematic investigation of interactions between papain and MPA-capped CdTe quantum dots, Mol. Biol. Rep., 2013, 40, 5781–5789 CrossRef CAS PubMed.
- Q. Xiao, S. Huang, J. Q. Ma, W. Su, P. Y. Li, J. G. Cui and Y. Liu, Systematically investigation of interactions between BSA and different charge-capped CdSe/ZnS quantum dots, J. Photochem. Photobiol., A, 2013, 249, 53–60 CrossRef PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 3rd edn, 2006 Search PubMed.
- V. P. Bogoeva, M. A. Radeva, L. Y. Atanasova, S. R. Stoitsova and R. N. Boteva, Fluorescence analysis of hormone binding activities of wheat germ agglutinin, Biochim. Biophys. Acta, 2004, 1698, 213–218 CrossRef CAS PubMed.
- M. A. Jhonsi, A. Kathiravan and R. Renganathan, Spectroscopic studies on the interaction of colloidal capped CdS nanoparticles with bovine serum albumin, Colloids Surf., B, 2009, 72, 167–172 CrossRef PubMed.
- C. J. Wilson and R. A. Copeland, Spectroscopic characterization of arrestin interactions with competitive ligands: study of heparin and phytic acid binding, J. Protein Chem., 1997, 16, 755–763 CrossRef CAS.
- C. B. Murphy, Y. Zhang, T. Troxler, V. Ferry, J. J. Martin and W. E. Jones, Probing Förster and Dexter energy-transfer mechanisms in fluorescent conjugated polymer chemosensors, J. Phys. Chem. B, 2004, 108, 1537–1543 CrossRef CAS.
- R. M. Watt and E. W. Voss, Solvent perturbation of the fluorescence of fluorescein bound to specific antibody, J. Biol. Chem., 1979, 254, 1684–1690 CAS.
- D. Leckband, Measuring the forces that control protein interactions, Annu. Rev. Biophys. Biomol. Struct., 2000, 29, 1–26 CrossRef CAS PubMed.
- D. P. Ross and S. Subramanian, Thermodynamics of protein association reactions: forces contributing to stability, Biochemistry, 1981, 20, 3096–3102 CrossRef.
- Y. J. Hu, Y. Liu, W. Jiang, R. M. Zhao and S. S. Qu, Fluorometric investigation of the interaction of bovine serum albumin with surfactants and 6-mercaptopurine, J. Photochem. Photobiol., B, 2005, 80, 235–242 CrossRef CAS PubMed.
- S. F. Sun, B. Zhou, H. N. Hou, Y. Liu and G. Y. Xiang, Studies on the interaction between oxaprozin-E and bovine serum albumin by spectroscopic methods, Int. J. Biol. Macromol., 2006, 39, 197–200 CrossRef CAS PubMed.
- Y. Li, W. Y. He, J. Q. Liu, F. L. Sheng, Z. D. Hu and X. G. Chen, Binding of the bioactive component Jatrorrhizine to human serum albumin, Biochim. Biophys. Acta, 2005, 1722, 15–21 CrossRef CAS PubMed.
- D. E. Epps, T. J. Raub and F. J. Kézdy, A general wide-range spectrofluorometric method for measuring the site-specific affinities of drugs toward human serum albumin, Anal. Biochem., 1995, 227, 342–350 CrossRef CAS.
- Y. J. Hu, Y. Liu and X. H. Xiao, Investigation of the interaction between berberine and human serum albumin, Biomacromolecules, 2009, 10, 517–521 CrossRef CAS PubMed.
- V. T. G. Chuang and M. Otagiri, Stereoselective binding of human serum albumin, Chirality, 2006, 18, 159–166 CrossRef CAS PubMed.
- T. Peters Jr, All About Albumin: Biochemistry, Genetics and Medical Applications, Academic Press, San Diego, 1996 Search PubMed.
- C. Long, E. J. King and W. M. Sperry, Biochemists' Handbook, E. & F. N. Spon Ltd, London, 1961 Search PubMed.
- B. Valeur, Molecular Fluorescence: Principles and Applications, Wiley Press, New York, 2001 Search PubMed.
- P. F. Qin, R. T. Liu, X. R. Pan, X. Y. Fang and Y. Mou, Impact of carbon chain length on binding of perfluoroalkyl acids to bovine serum albumin determined by spectroscopic methods, J. Agric. Food Chem., 2010, 58, 5561–5567 CrossRef CAS PubMed.
- F. Samari, B. Hemmateenejad, M. Shamsipur and M. Rashidi, Affinity of two novel five-coordinated anticancer Pt(II) complexes to human and bovine serum albumins: a spectroscopic approach, Inorg. Chem., 2012, 51, 3454–3464 CrossRef CAS PubMed.
- S. R. Feroz, S. B. Mohamad, N. Bujang, S. N. A. Malek and S. Tayyab, Multispectroscopic and molecular modeling approach to investigate the interaction of flavokawain B with human serum albumin, J. Agric. Food Chem., 2012, 60, 5899–5908 CrossRef CAS PubMed.
- G. W. Zhang, L. Wang and J. H. Pan, Probing the binding of the flavonoid diosmetin to human serum albumin by multispectroscopic techniques, J. Agric. Food Chem., 2012, 60, 2721–2729 CrossRef CAS PubMed.
- Z. X. Lu, T. Cui and Q. L. Shi, Application of circular dichroism and optical rotatory dispersion in molecular biology, Science Press, Beijing, 1st edn, 1987, pp. 79–82 Search PubMed.
- S. Li, Y. Z. Wang, J. G. Jiang and S. J. Dong, pH-Dependent protein conformational changes in albumin: gold nanoparticle bioconjugates: a spectroscopic study, Langmuir, 2007, 23, 2714–2721 CrossRef PubMed.
- Z. Y. Chen, H. Y. Xu, Y. L. Zhu, J. Y. Liu, K. Y. Wang, P. X. Wang, S. J. Shang, X. N. Li, Z. L. Wang, W. Shao and S. D. Zhang, Understanding the fate of an anesthetic, nalorphine upon interaction with human serum albumin: a photophysical and mass-spectroscopy approach, RSC Adv., 2014, 4, 25410–25419 RSC.
- S. Huang, F. W. Zhu, H. N. Qiu, Q. Xiao, Q. Zhou, W. Su and B. Q. Hu, A sensitive quantum dots-based “OFF–ON” fluorescent sensor for ruthenium anticancer drugs and ctDNA, Colloids Surf., B, 2014, 117, 240–247 CrossRef CAS PubMed.
- J. Zhang, R. Li, F. L. Jiang, B. Zhou, Q. Y. Luo, Q. L. Y. Yu, X. L. Han, Y. Lin, H. He, Y. Liu and Y. L. Wang, An electrochemical and surface plasmon resonance study of adsorption actions of DNA by Escherichia coli, Colloids Surf., B, 2014, 117, 68–74 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |