DOI:
10.1039/C4RA06064D
(Paper)
RSC Adv., 2014,
4, 38182-38191
High nuclearity Ni(II) cages from hydroxamate ligands†
Received
21st June 2014
, Accepted 13th August 2014
First published on 13th August 2014
Abstract
The synthesis, structural and magnetic characterisation of a family of Ni(II) cages built from hydroxamate ligands is presented. Two pentanuclear 12-MCNi(II)-4 metallacrowns [Ni5(L1)4(MeOH)4](ClO4)2·2MeOH (1) and [Ni5(L1)4(py)5](ClO4)2·H2O (2) (where L1H2 = 2-(dimethylamino)phenylhydroxamic acid) share analogous, near-planar {Ni5(L1)4}2+ cores, but differ in the number and nature of the ligands located at the axial Ni(II) sites; the addition of pyridine converting square planar Ni(II) ions to square-based pyramidal and octahedral Ni(II) ions, introducing extra paramagnetic metal centres which ‘switch on’ additional magnetic superexchange pathways. Subtle variations in the reaction scheme used to produce complexes 1 and 2 result in both a change of topology and an increase in nuclearity, through isolation of the hepta- and nonametallic complexes [Ni7(L1H)8(L1)2(H2O)6](SO4)·15H2O (3), [Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](SO4)·29H2O (4) and [Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](ClO4)2·2MeOH·18H2O (5) (where L2H2 = 2-(amino)phenylhydroxamic acid). Complementary dc magnetic susceptibility studies and DFT analysis indicate dominant antiferromagnetic exchange interactions in 1, 2, 4 and 5, but competing ferro- and antiferromagnetic exchange in 3.
Introduction
The role of hydroxamic acids (of general formula RCONHOH; Scheme 1) in biology and bioinorganic chemistry cannot be underestimated due to their rich bioactivity originating from their inherent pharmacological, toxicological and pathological properties.1,2 More specifically these organic acids are able to act as efficient siderophores as well as effective selective enzyme inhibitors for histone deacetylase, ureases and prostaglandin H2 synthases.1 Such behaviour stems from their ability to bind strongly to numerous transition metal centres, rendering the catalytic active site impotent in the process and are therefore important ingredients in numerous therapeutic drugs.1,2 This chelating ability is also the reason that hydroxamic acids have also been shown to act as effective ligands in the field of coordination chemistry,3 while industrial application lies in their use in the extraction and subsequent recovery of numerous transition metals (Fe, Co, Ni, Cu, Zn and Cd).4
 |
| | Scheme 1 Generic molecular structure of the hydroxamic acid ligands used in this work (R = Me; L1H2; R = H; L2H2). | |
Our own work using the ligands 2-(dimethylamino)phenylhydroxamic acid (L1H2) and 2-(amino)phenylhydroxamic acid (L2H2) has led to the synthesis of a family of pentametallic 12-MCCu(II)-4 metallacrowns,5 whose {Cu5(Lx)4}2+ (x = 1, 2) cores could subsequently undergo ligand addition and substitution in a controlled manner towards the premeditated formation of 1- and 2-D extended networks comprising {Cu5} metallacrown nodes.6 Herein we present an addition to this work, with the synthesis, structures and magnetic characterisation of a family of novel Ni(II) cages of varying nuclearities and topologies. The 12-MCNi(II)-4 metallacrowns [Ni5(L1)4(MeOH)4](ClO4)2·2MeOH (1) and [Ni5(L1)4(py)5](ClO4)2·H2O (2) have similar cores, but differ in the number and nature of ligands bonded to the axial sites on the Ni(II) ions, with addition of pyridine converting square planar (s = 0) Ni(II) ions in 1 to square-based pyramidal/octahedral Ni(II) ions (s = 1) in 2, introducing additional magnetic superexchange interactions to be “switched on”. Variation in reactions conditions (metal salt, ligand type, base, solvent) leads to both a change in topology to non-metallacrown cages, and an increase in nuclearity from penta- to hepta- and nonametallic.
Results and discussion
Complex 1 is produced upon reaction of Ni(ClO4)2·6H2O, L1H2 and NaOH in MeOH (see Experimental section for full details), while 2 is made by simply adding pyridine to the same reaction. Systematic variation of the amount of pyridine used did not affect the identity of the isolate product, nor the number of bound pyridine ligands. Complexes 1 and 2 crystallise in the triclinic space group P![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) and monoclinic space group P21/n, respectively. Pertinent crystallographic data is given in Table 1. Complexes 1 and 2 (Fig. 1) contain near planar {Ni5(L1)4}2+ cores, with the central Ni(II) ions (labelled Ni1 in both cases) surrounded by an outer ring or wheel of four Ni(II) centres (Ni2, Ni3 and symmetry equivalent (s.e.) in 1 and Ni2–Ni5 in 2), themselves connected into a 12-MC-4 metallacrown via four doubly deprotonated L12− ligands displaying a η1:η2:η1:η1,μ3-bonding motif (Fig. 1 and S1†). Although analogous in many ways, important structural differences between 1 and 2 lie in the coordination geometries at the metal centres. The central Ni(II) ion in 1 exhibits distorted octahedral geometry, possessing two axially ligated MeOH ligands (Ni1–O5 = 2.153 Å). Two of the four outer metal centres (Ni3 and s.e.) are five coordinate and square based pyramidal, with τ = 0.08,7 due to the presence of a single axially bound MeOH ligand (Ni3–O6 = 2.036 Å). The remaining two outer Ni(II) centres (Ni2 and s.e.) are not axially ligated, and thus are four coordinate and square planar in geometry. The addition of pyridine leads to different coordination at the Ni(II) centres in 2. Firstly the central nickel adopts a distorted square-based pyramidal configuration (τ = 0.34) with one axially bound pyridine ligand (Ni1–N13 = 2.012 Å). Likewise the outer ions Ni2 and Ni3 exhibit distorted square pyramidal geometries (τ = 0.34 and 0.15, respectively), each with one terminal pyridine ligand (Ni2–N3 = 2.029 Å, Ni3–N6 = 2.036 Å). Ni5 is the only six coordinate metal ion, possessing both axially and equatorially bound pyridine ligands (Ni5–N10 = 2.135 Å and Ni5–N11 = 2.090 Å respectively). The effect of this additional pyridine coordination is that the adjacent L12− ligand significantly distorts away from the {Ni5} plane, forcing it to bond at the axial Ni5 site via its –NMe2 group (Fig. 1). The outer Ni4 ion remains in a four coordinate square planar geometry, suggesting it to be the sole diamagnetic metal centre in 2 (vide infra). The axial pyridine ligands coordinated to Ni1, Ni3 and Ni5 appear almost superimposable when viewed along the plane of the molecule, lying at distances typical of πcentroid–πcentroid interactions ([C52–N10]⋯[C50–N13] = 3.674 Å and [C50–N13]⋯[C42–N6] = 3.651 Å) (Fig. 1d). Upon close scrutiny of 1 and 2 it becomes apparent that pyridine ligation has promoted a puckering of the {Ni5} core in 2 when compared to the near planar pentametallic skeleton in 1 (Fig. 1b cf. 1d).
and monoclinic space group P21/n, respectively. Pertinent crystallographic data is given in Table 1. Complexes 1 and 2 (Fig. 1) contain near planar {Ni5(L1)4}2+ cores, with the central Ni(II) ions (labelled Ni1 in both cases) surrounded by an outer ring or wheel of four Ni(II) centres (Ni2, Ni3 and symmetry equivalent (s.e.) in 1 and Ni2–Ni5 in 2), themselves connected into a 12-MC-4 metallacrown via four doubly deprotonated L12− ligands displaying a η1:η2:η1:η1,μ3-bonding motif (Fig. 1 and S1†). Although analogous in many ways, important structural differences between 1 and 2 lie in the coordination geometries at the metal centres. The central Ni(II) ion in 1 exhibits distorted octahedral geometry, possessing two axially ligated MeOH ligands (Ni1–O5 = 2.153 Å). Two of the four outer metal centres (Ni3 and s.e.) are five coordinate and square based pyramidal, with τ = 0.08,7 due to the presence of a single axially bound MeOH ligand (Ni3–O6 = 2.036 Å). The remaining two outer Ni(II) centres (Ni2 and s.e.) are not axially ligated, and thus are four coordinate and square planar in geometry. The addition of pyridine leads to different coordination at the Ni(II) centres in 2. Firstly the central nickel adopts a distorted square-based pyramidal configuration (τ = 0.34) with one axially bound pyridine ligand (Ni1–N13 = 2.012 Å). Likewise the outer ions Ni2 and Ni3 exhibit distorted square pyramidal geometries (τ = 0.34 and 0.15, respectively), each with one terminal pyridine ligand (Ni2–N3 = 2.029 Å, Ni3–N6 = 2.036 Å). Ni5 is the only six coordinate metal ion, possessing both axially and equatorially bound pyridine ligands (Ni5–N10 = 2.135 Å and Ni5–N11 = 2.090 Å respectively). The effect of this additional pyridine coordination is that the adjacent L12− ligand significantly distorts away from the {Ni5} plane, forcing it to bond at the axial Ni5 site via its –NMe2 group (Fig. 1). The outer Ni4 ion remains in a four coordinate square planar geometry, suggesting it to be the sole diamagnetic metal centre in 2 (vide infra). The axial pyridine ligands coordinated to Ni1, Ni3 and Ni5 appear almost superimposable when viewed along the plane of the molecule, lying at distances typical of πcentroid–πcentroid interactions ([C52–N10]⋯[C50–N13] = 3.674 Å and [C50–N13]⋯[C42–N6] = 3.651 Å) (Fig. 1d). Upon close scrutiny of 1 and 2 it becomes apparent that pyridine ligation has promoted a puckering of the {Ni5} core in 2 when compared to the near planar pentametallic skeleton in 1 (Fig. 1b cf. 1d).
Table 1 Crystallographic data for complexes 1, 2, 4 and 5
| |
1·2MeOH |
2·H2O |
4·29H2O |
5·2MeOH·18H2O |
| Includes guest molecules. Mo-Kα radiation, graphite monochromator. wR2 = [Σw(|Fo2| − |Fc2|)2/Σw|Fo2|2]1/2. For observed data. R1 = Σ||Fo| − |Fc||/Σ|Fo|. |
| Formulaa |
C42H64N8O22Cl2Ni5 |
C61H67N13O17Cl2Ni5 |
C70H130N20O57S1Ni9 |
C74H116N20O52Cl2Ni9 |
| MW |
1397.46 |
1618.71 |
2724.19 |
2716.96 |
| Crystal system |
Triclinic |
Monoclinic |
Orthorhombic |
Monoclinic |
| Space group |
P |
P21/n |
Ima2 |
Cc |
| a/Å |
11.191(2) |
14.6573(4) |
29.1847(11) |
20.0036(7) |
| b/Å |
12.389(3) |
15.1811(4) |
21.2385(7) |
25.0628(9) |
| c/Å |
12.401(3) |
29.7812(11) |
19.7536(6) |
21.2234(5) |
| α/° |
70.12(3) |
90 |
90 |
90 |
| β/° |
63.48(3) |
93.552(3) |
90 |
90.320(2) |
| γ/° |
64.17(3) |
90 |
90 |
90 |
| V/Å3 |
1362.0(5) |
6614.0(4) |
12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 244.1(7) 244.1(7) |
10640.1(6) |
| Z |
1 |
4 |
4 |
4 |
| T/K |
150(2) |
150(2) |
150(2) |
150(2) |
| λb/Å |
0.7107 |
0.7107 |
0.7107 |
0.7107 |
| Dc/g cm−3 |
1.704 |
1.624 |
1.192 |
1.656 |
| μ(Mo-Kα)/mm−1 |
1.880 |
1.558 |
1.431 |
1.708 |
| Collected/unique, (Rint) refl. |
10303/4974, (0.0169) |
52717/12096, (0.0785) |
52181/11376, (0.1403) |
40644/16130, (0.0946) |
| wR2 (all data) |
0.0758 |
0.11398 |
0.1531 |
0.1769 |
| R1d,e |
0.0308 |
0.0482 |
0.0844 |
0.0693 |
| Goodness of fit on F2 |
1.024 |
1.023 |
0.964 |
1.027 |
| Flack parameter |
n.a |
n.a |
0.009(18) |
0.026(18) |
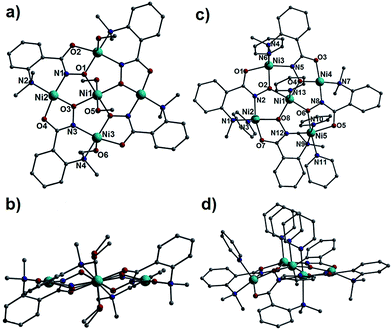 |
| | Fig. 1 Crystal structures of 1 and 2 viewed perpendicular (a and c) and parallel (b and d) to their {Ni(II)5} planar cores. Colour code: light blue (Ni), red (O), dark blue (N), grey (C). The perchlorate counter anions have been omitted for clarity. Hydrogen atoms have been omitted for clarity in all cases. | |
The {Ni5(L1)4(MeOH)4}2+ (1) and {Ni5(L1)4(py)5}2+ (2) cations are each charge balanced by two ClO4− counter anions, sitting above and below the planar {Ni5} array in 1 and at the periphery of the structure in 2, the difference presumably due to the presence of the coordinated pyridine ligands (Fig. 2) and subsequent changes to intermolecular interactions. In both cases the O donor atoms of the ClO4− counter anions forge intermolecular H-bonding interactions with nearby {Ni5} units in all three directions. More specifically the ClO4− units in 1 hydrogen bond via aromatic (e.g. C12(H12)⋯O8 = 2.506 Å) and aliphatic protons belonging to nearby hydroxamate and terminally bonded MeOH molecules, respectively (e.g. O5(H5H)⋯O10 = 2.445 Å). Hydrogen bonding is also observed between the terminal and interstitial MeOH molecules (e.g. O6(H6H)⋯O11 = 2.230 Å). In 2 the predominant H-bonding occurs between the ClO4− counter anions and aromatic hydroxamate protons (C25(H25)⋯O13 = 2.584 Å, C40(H40)⋯O16 = 2.590 Å and C43(H43)⋯O14 = 2.585 Å). The {Ni5} units in 1 pack in superimposable columns along the a cell direction and these stacks are connected through π–π stacking interactions between adjacent hydroxamate aromatic rings ([C2–C7]centroid⋯[C2′–C7′]centroid = 3.897 Å; Fig. 2, left). The pentametallic cages in 2 arrange themselves into 2D brickwork sheets along the ab cell diagonal, with these sheets lying in superimposable rows down c, as shown in Fig. 2.
 |
| | Fig. 2 Packing arrangements of 1 (left) and 2 (right) as viewed down the b axis of their respective unit cells. Hydrogen atoms have been omitted for clarity. Perchlorate counter anions are represented in the space-fill mode. | |
Despite numerous attempts we could not produce the analogous metallacrowns to 1 and 2 using L2H2. The formation of 1 and 2 adds to the relatively small number of 12-MCNi(II)-4 metallacrowns known in the literature,8 and are the first constructed using 2-(dimethylamino)phenylhydroxamic acid (L1H2). Interestingly the general 12-MCNi(II)-4 framework also appears as a building block within the elaborate and rather unusual fused metallacrown dimer Ni(II)2(mcpa)2(CH3OH)3(H2O)[12-MCNi(II)N(shi)2(pko)2-4][12-MCNi(II)N(shi)3(pko)-4] (where Hmpca = 2-methyl-4-chlorophenoyacetic acid, Hpko = di-(2-pyridyl)ketone oxime and H3shi = salicylhydroxamic acid).9
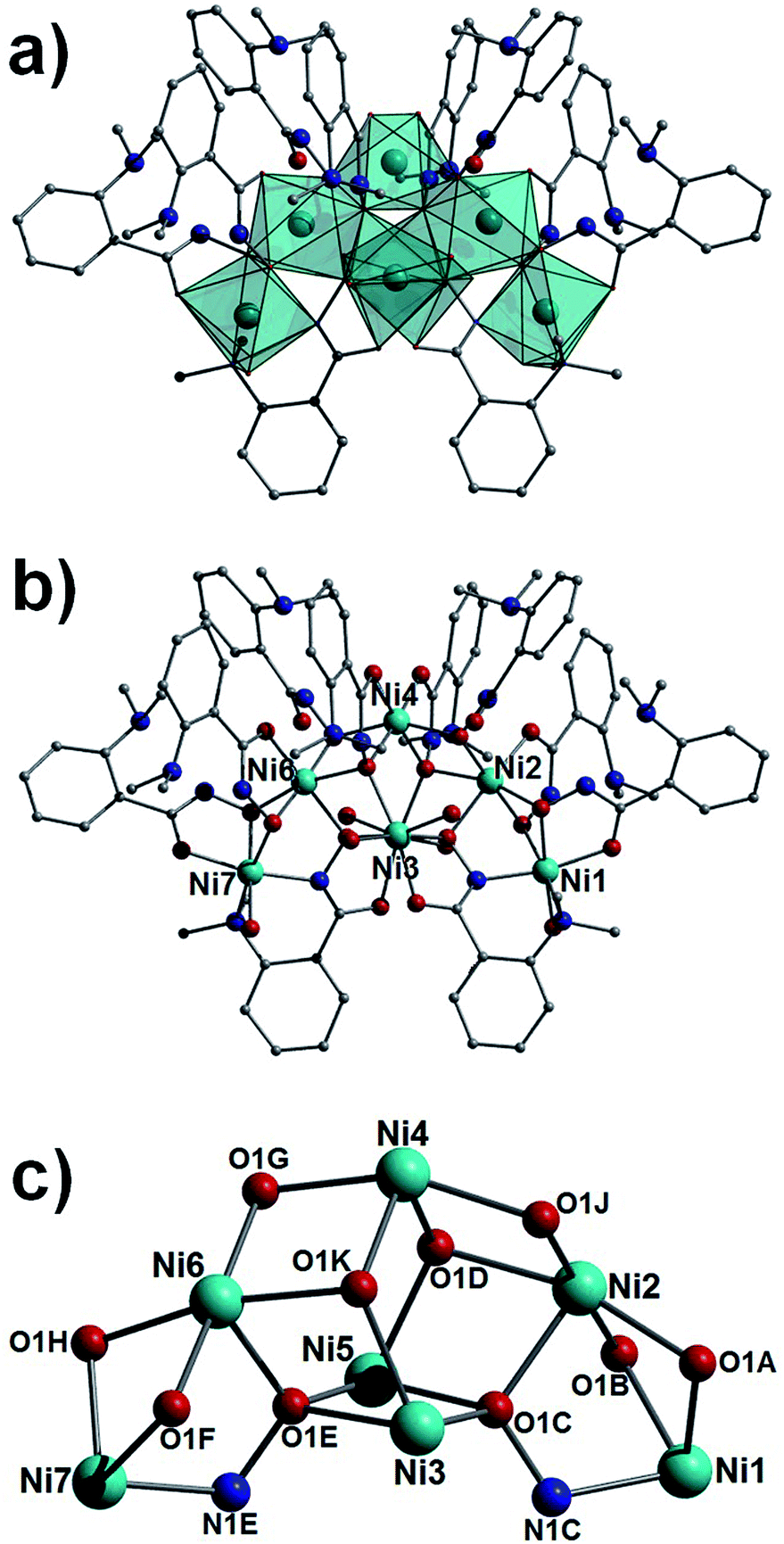
As previously communicated by one of us,10 the reaction of NiSO4·6H2O, L1H2 in a basic solvent mixture of H2O and MeOH gives the heptanuclear complex [Ni7(L1H)8(L1)2(H2O)6](SO4)·15H2O (3), a complex whose structure deviates significantly from the metallacrowns of 1 and 2. The core in 3 (Fig. 3) shows a trigonal bipyramidal array (or alternatively two face-sharing tetrahedra) of nickel(II) ions (Ni2–Ni6) with an extra two metal centres annexed at the apical sites (Ni1 and Ni7). All nickel centres exhibit distorted octahedral geometries and are connected through hydroxamate ligands showing four types of bonding mode (η2:μ-, η1:η3:μ3-, η1:η2:μ- and η1:η3:η1:η1:μ4-; Fig. 3 and S1†). Terminal water molecules complete the coordination spheres at Ni1 and Ni7 (Ni1–O1 = 2.074 Å, Ni7–O5 = 2.090 Å), and at the equatorial Ni3 and Ni5 sites. Intramolecular πcentroid–πcentroid interaction at distances of (Å) 3.829 ([C2B–C7B]⋯[C2D–C7D]) and 3.991 ([C2F–C7F]⋯[C2K–C7K]) exist between aromatic rings of nearest neighbour hydroxamate ligands. A single charge balancing SO42− counter anion lies at the periphery of the structure, H-bonding to protons of metal bound H2O ligands (e.g. O4(H4A)⋯O3SS = 2.005 Å) and waters of crystallisation (e.g. O21(H21A)⋯O1SS = 1.875 Å). In the crystal the individual {Ni7} moieties in 3 arrange into superimposable rows along the a cell direction, packing in a brickwork topology in the bc plane (Fig. S2†).
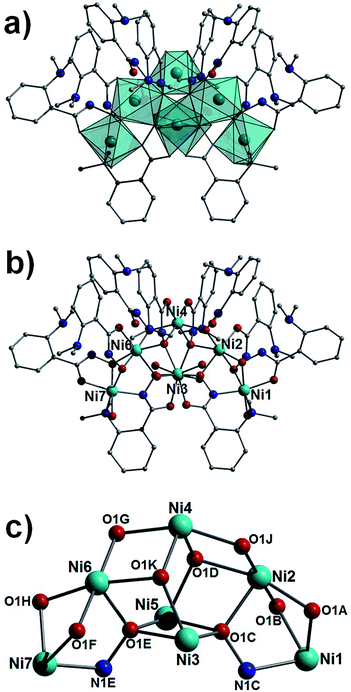 |
| | Fig. 3 Polyhedral (a) and regular (b) representation of the crystal structure in 3. (c) Metallic core in 3. Colour code as in Fig. 1. Hydrogen atoms have been omitted for clarity. | |
The reaction of NiSO4·6H2O and L2H2 in a basic MeOH–H2O solution afforded an even larger cage, [Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](SO4)·29H2O (4). Moreover the perchlorate salt of 4, [Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](ClO4)2·2MeOH·18H2O (5) was readily produced using a similar synthetic procedure (see Experimental section for details). Complexes 4 (Fig. S3†) and 5 (Fig. 4) crystallise in the orthorhombic Ima2 and monoclinic Cc space groups, respectively. Pertinent crystallographic details are given in Table 1. The cores in 4 and 5 are best described as comprising two tetrahedral arrays of distorted octahedral Ni(II) ions linked by a single, central, six coordinate Ni(II) metal centre (labelled Ni5 in both cases). The Ni(II) ions are connected by a combination of four singly (LH−) and six doubly (L2−) deprotonated hydroxamate ligands exhibiting η1:η2, μ- and η1:η3:η1:η1, μ4-bonding modes, respectively (Fig. S1†). In both cases two μ-bridging H2O ligands connect the central Ni5 ion to the tetrahedral units, while terminal water molecules complete the coordination spheres at the two peripheral Ni(II) centres (Ni3–O11 = 2.022 Å in 4; Ni1–O3 = 2.051 Å and Ni9–O17 = 2.042 Å in 5). The resultant {Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2}2+ cationic cages are charge balanced by one SO42− and two ClO4− counter anions, respectively.
 |
| | Fig. 4 Polyhedral (a) and regular (b) representation of the crystal structure in 5. (c) The metallic core in 5. Colour code as used in Fig. 1 and elsewhere in the text. The majority of H atoms have been omitted for clarity however the –NH2 protons are represented as black spheres. | |
The individual {Ni9} units in 4 arrange in the common brickwork motif along the bc plane (Fig. 5, left) and are connected to one another via πcentroid–πcentroid stacking interactions between hydroxamate aromatic rings of neighbouring {Ni9} units ([C8–C13]⋯[C15–C20] = 3.605 Å). These sheet-like arrangements stack in superimposable rows along the a cell direction to complete the topology in 4 in the crystal (Fig. 5, left). The [Ni9] cages in 5 also arrange themselves in the brickwork motif along the ac plane of the unit cell, these 2D sheets stack in a staggered arrangement as opposed to the superimposable rows observed in 4 (Fig. S4†). In a similar fashion to 4, πcentroid–πcentroid stacking interactions connect the individual [Ni9] nodes in the brickwork topology ([C9–C14]⋯[C58–C63] = 3.538 Å) and this is aided by numerous H-bonding interactions between aliphatic protons of the hydroxamate ligands (–NH2 and = NH groups) and waters of crystallisation-effectively acting as molecular mortar in the packing in 5 (i.e. N17(H17B)⋯O28 = 2.386 Å).
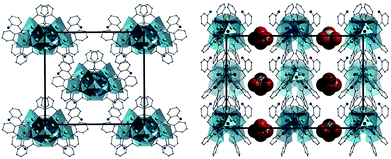 |
| | Fig. 5 Crystal packing observed in 4 as viewed along the a (left) and b (right) cell direction. All hydrogen atoms and solvents of crystallisation have been omitted for clarity. The SO4− counter anions are represented as space-fill in the figure on the right and removed for clarity on the left. | |
It is somewhat difficult to rationalise the change in structure from 1 and 2 (Ni5) to 3 (Ni7), since the reactions involve the use of a different metal salt (perchlorate versus sulphate) and different solvent (MeOH versus MeOH–H2O). The difference in reaction schemes between [Ni5] and [Ni9] involve a change in ligand, base and solvent, while the difference in the reaction that produces [Ni7] versus [Ni9] is a change in ligand and base. Elucidating the roles of each reaction variable would therefore require a larger library of complexes to be isolated, and we are currently working to that end. However we can say that the role of ligand selection (i.e. L12− in 3 versus L22− in 4) and more specifically functional group dictated steric effects (Me groups in L12− versus H groups L22−) on producing complex 4 over 3 cannot be ignored in terms of structure-directing influences.
Theoretical determination of paramagnetic centres in complexes 1 and 2
Complexes 1 and 2 each possess five Ni(II) ions displaying a total of three different [distorted] geometries – square planar, square-based pyramidal and octahedral. Square planar Ni(II) ions are generally diamagnetic, whereas square-based pyramidal Ni(II) ions can be either diamagnetic or paramagnetic depending on the axial ligand strength.11 Ni(II) ions in octahedral environments on the other hand are always paramagnetic in nature. To confirm the electronic structure of these metal ions we have performed DFT calculations employing varying combinations of spin states (s = 0 versus s = 1) at each of the Ni(II) centres in 1 and 2 (Fig. 6). We began by assuming that all the Ni(II) ions in complex 1 were paramagnetic (s = 1) and then gradually decreased the number of paramagnetic centres while increasing the number of diamagnetic (s = 0) metal ions. Five different spin states have been computed for 1 giving rise to triplet ground states for Ni1 (octahedral), Ni3 and Ni3A (square pyramidal). All other configurations outlined in Fig. 6 lie much higher in energy compared to our calculated ground spin state configuration and thus are unlikely to be accessible at ambient conditions; that is, square planar Ni2 and Ni2A have isolated s = 0 ground states and we can conclude that all experimental magnetic properties in 1 are exclusively due to paramagnetic (s = 1) ground state configurations at the Ni1, Ni3 and Ni3A positions. In a similar vein, five different configurations were computed for complex 2. Here, it was found that Ni(II) centres Ni1, Ni2, Ni3 and Ni5 are paramagnetic (s = 1) and square planar Ni4 diamagnetic (s = 0), with all other possible configurations lying higher in energy. However unlike complex 1, these excited state configurations lie somewhat closer than in 1, with the first excited state lying 42 kJ mol−1 above the ground state (Fig. 6, right).
 |
| | Fig. 6 All possible total spin (s) configurations of the individual Ni(II) ions in complexes 1 and 2, with their respective energies (kJ mol−1). | |
Magnetic susceptibility measurements
Dc magnetic susceptibility measurements were performed on powdered microcrystalline samples of 1–3 and 5 in the 300–5 K temperature range, in an applied field of 0.1 T, and are plotted as their χMT products in Fig. 7. For the pentametallic species 1 and 2, the room temperature χMT values of 3.55 cm3 mol−1 K (1) and 3.43 cm3 mol−1 K (2) are below that expected for three (3.63 cm3 mol−1 K) and four (4.84 cm3 mol−1 K) non interacting, paramagnetic Ni(II) centres, assuming g = 2.2. Both show a steady decrease in χMT upon decreasing temperature (although the curve in 2 is steeper than that in 1), reaching values of 1.28 and 1.10 cm3 mol−1 K at 5 K, respectively. This is indicative of the presence of dominant intramolecular antiferromagnetic interactions in both complexes, with the exchange in 2 being somewhat stronger than in 1.
 |
| | Fig. 7 Plot of χMT vs. T for complexes 1 (□), 2 (○), 3 (◊) and 5 (△). The solid lines represent best-fits of the experimental data. See text for details. | |
For the interpretation of the magnetic properties of 1 and 2 we employed the models given in Fig. 8. Here, J1 is the isotropic exchange interaction parameter between the central Ni ion and the paramagnetic ions that surround it, mediated by one Ni–O–Ni and one Ni–O–N–Ni interaction; J2 describes the interaction around the outer ‘wheel’ between the peripheral Ni ions, mediated by one Ni–O–N–Ni interaction.12 The best-fit parameters obtained were J1 = −3.51 cm−1 (1) and J1 = −16.87 cm−1 and J2 = −7.83 cm−1 (2). The ground state of 1 is an S = 1 state, and the ground state in 2 is also an S = 1 state, but with an S = 0 state just 1.43 cm−1 above (Fig. 9). The individual Ni–Ooximato–Ni magnetic pathways in 1 (Ni1–O1–Ni3 = 103.98°) and 2 (Ni1–O8–Ni2 = 113.85°, Ni1–O2–Ni3 = 120.54° and Ni1–O6–Ni5 = 115.18°) each lie in the range expected for mediating antiferromagnetic exchange, with larger angles mediating stronger interactions as observed experimentally.13,14
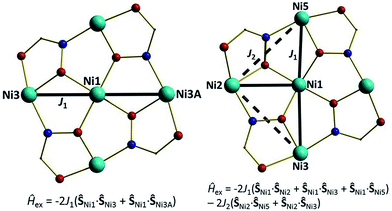 |
| | Fig. 8 Schematic illustrating the models used to fit the experimental data for complexes 1 and 2. See main text for fitting parameters. | |
 |
| | Fig. 9 Energy versus total spin state for the lowest lying S states in 1 and 2 as determined from the isotropic fit of the susceptibility data. | |
Magnetic susceptibility studies on complex 5 also show the presence of dominant antiferromagnetic exchange between the Ni(II) centres (Fig. 7), but the data for complex 3 suggests more competition between ferro- and antiferromagnetic exchange. The room temperature χMT values of 7.87 and 7.98 cm3 mol−1 K, respectively, are below the values expected for seven and nine non-interacting paramagnetic Ni(II) ions (8.47 (3) and 10.89 (5) cm3 mol−1 K, assuming g = 2.2). For complex 5, the value decreases monotonically with decreasing temperature, reaching 1.14 cm3 mol−1 K at T = 5 K. The variable T data for 3 are a little more complex. The χMT product decreases steadily but slowly to approximately T = 25 K where it then plateaus at a value of ∼4.5 cm3 mol−1 K, before decreasing again at lower temperatures, reaching a minimum value of 3.95 cm3 mol−1 K. The structural complexity of 3 and 5 precludes detailed quantitative analyses of the susceptibility data, since there are numerous different exchange interactions. However, the magnitude of the exchange can be estimated through the employment of simple models. In each case we attempted to fit the susceptibility with just one J value, assuming all Ni⋯Ni interactions to be of similar magnitude. This approach was successful for complex 5 and afforded J1 = −5.27 cm−1 with g fixed to 2.2. For complex 3, this approach did not work and two J values were required (Fig. S5†); one (J1) to describe Ni ions connected by a one-atom (Ni–O–Ni) bridge, and one (J2) to describe Ni ions connected by two-atom (Ni–O–N–Ni) bridges. This afforded the best fit parameters J1 = +0.64 cm−1 and J2 = −8.94 cm−1 (3) with g fixed to 2.2. These numbers are a guide only, but are similar to structurally similar Ni(II) cages previously reported in the literature.13,14
Theoretical studies of the magnetic exchange in complexes 1 and 2
DFT studies were carried out to compute the intramolecular magnetic exchange coupling in complexes 1 and 2. More specifically we computed the energies of four different spin configurations to obtain two exchange coupling constants corresponding to the ground state in 1 (see Table S1† for all computed configurations). Since complex 2 is asymmetric, five independent J values were computed using seven different spin configurations (see Table S2† for all computed configurations). The corresponding Hamiltonians for 1 and 2 are given in the computational details section and all computed magnetic coupling constants calculated for complex 1 and 2 are shown in Fig. 10.
 |
| | Fig. 10 Schematic representation of the magnetic coupling constants (in cm−1) in 1 (left) and 2 (right). | |
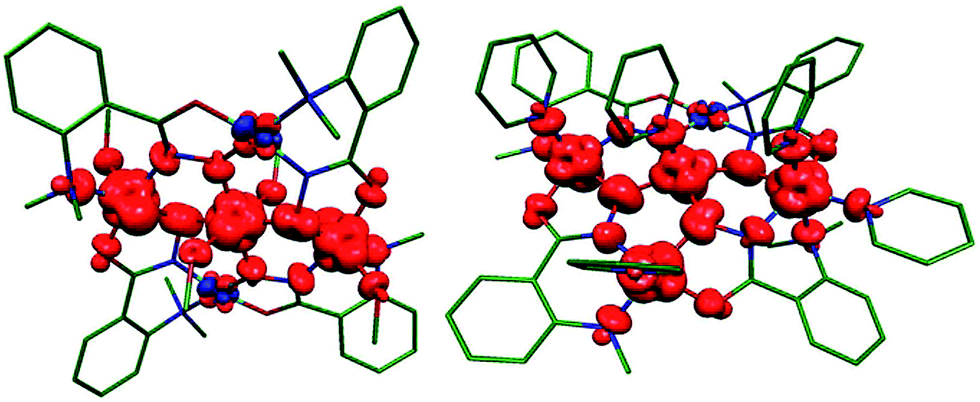
Calculations on complex 1 yielded weak antiferromagnetic J values (J1A = J1B = −0.4 cm−1) in agreement with those obtained experimentally, albeit somewhat smaller in magnitude. The computed spin density plot for the high spin configuration (an S = 3 state) in 1 is shown in Fig. 11 and clearly shows dominant spin delocalization leading to AF coupling. Computed overlap integrals support this argument where significant overlap between dx2–y2 orbitals is detected (see Table S3† for details).
 |
| | Fig. 11 DFT computed spin density plots for complex 1 (left) and 2 (right). Here red and blue indicates positive and negative spin densities respectively. | |
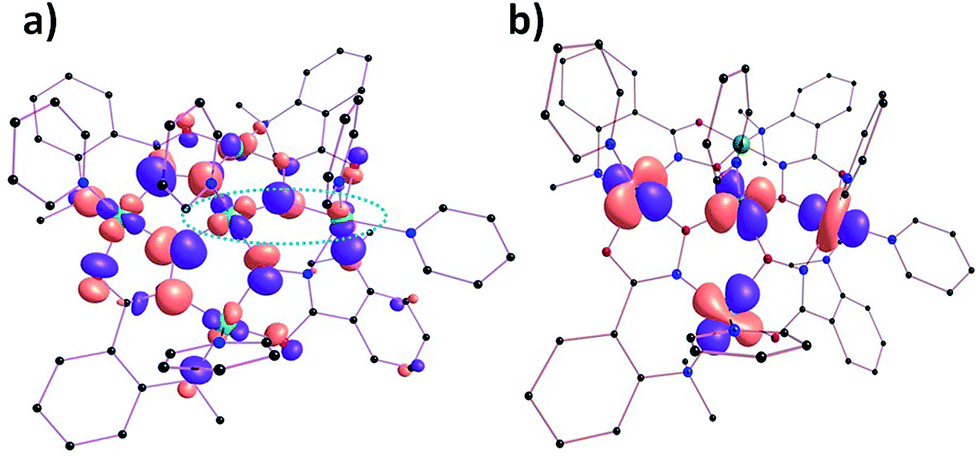
Calculations carried out on complex 2 again reveal that J2 = J2A = J2B and that the experimental (J2 = −7.83 cm−1) and theoretical (J2A = −9.5 cm−1 and J2B = −10.0 cm−1) values are very similar in magnitude. The computed J1 values (J1A–C) suggest the presence of a range of exchange types, from weakly ferromagnetic (J1C = +2.0 cm−1) to strongly antiferromagnetic (J1A = −20.2 cm−1), somewhat in contrast to that derived from experiment (J1 = −16.37 cm−1). The J1A–C exchange interactions in complex 2 are mediated via a combination of Ni–Ooximato–Ni and Ni–N–O–Ni moieties, while the J2A and J2B are mediated via NO bridges only. Interestingly all calculated J values were shown to be AF in nature apart from (ferromagnetic) J1C, which arises from the inherent orthogonality of the dx2–y2 orbitals belonging to Ni1 and Ni5. The orthogonality arises from the relatively acute Ni1–O6–Ni5 angle (115.18°) and the large Ni1–O8–N12–Ni5 dihedral twist (31.58°) observed along the Ni1⋯Ni5 pathway (Fig. 12 and Table S5†).
 |
| | Fig. 12 (a) One of the MOs of complex 2 highlighting orthogonality between the dx2–y2 magnetic orbitals of centres Ni1 and Ni5 respectively (circled). (b) Natural hybrid orbitals15 representing the dominant orbital interactions in the molecular plane of complex 2 (see ESI Table S4† for computed overlap integral values). | |
The dominant magnetic interaction in both [Ni5] complexes occurs in the plane of the molecule, and the relative (albeit small) differences in the exchange interactions between complexes 1 and 2 can be explained and visualised through the orientations of their molecular orbitals. While all paramagnetic Ni(II) ions in complex 1 have their dx2–y2 orbitals in the {Ni5} plane, Ni5 in complex 2 does not; here the dz2 orbital lies in the plane (Fig. 12b). The AF magnetic pathways in 2 arise because of significant overlap between the dx2–y2 and dx2–y2/dz2 magnetic orbitals as shown in Fig. 12b. Likewise, a significant dx2–y2|p|dx2–y2 overlap along the Ni1⋯Ni2 vector was detected, supporting a strong antiferromagnetic J1A (see Table S4†). The spin density plot shown in Fig. 11 reveals that a dominant spin delocalization mechanism is operative in 2, with larger spin densities residing on the O-atoms.
Concluding remarks
The hydroxamic acids 2-(dimethylamino)phenylhydroxamic acid (L1H2) and 2-(amino)phenylhydroxamic acid (L2H2) have been successfully used as bridging ligands in the synthesis of a family of Ni(II) cages ranging from penta- to nonametallic. Addition of pyridine to the reaction mixture that produces the 12-MCNi(II)-4 metallacrown [Ni5(L1)4(MeOH)4](ClO4)2·2MeOH (1) leads to additional axial ligation at some Ni(II) centres, forming the related metallacrown [Ni5(L1)4(py)5](ClO4)2·1H2O (2), converting square planar sites to square-based pyramidal/octahedral sites, ‘switching on’ additional magnetic superexchange pathways. DFT analysis computed triplet s = 1 ground states for the latter two geometries. Structural rearrangement to non-metallacrown topologies can be achieved through simple variation in reaction conditions, leading to the formation of the hepta and nonametallic cages [Ni7(L1H)8(L1)2(H2O)6](SO4)·15H2O (3), [Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](SO4)·29H2O (4) and [Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](ClO4)2·2MeOH·18H2O (5), which display metallic skeletons comprising two bi-capped, face-sharing tetrahedra in 3 and two annexed tetrahedra in 4 and 5. The linear arrangement of three antiferromagnetically coupled Ni(II) centres in 1 leads to a S = 1 ground state. Dominant antiferromagnetic exchange is also present in complexes 2 and 5, but competing ferro- and antiferromagnetic exchange between the seven nickel centres in complex 3 leads to the stabilisation of an intermediate ground spin state. DFT calculations were employed to ascertain the electronic structure of the Ni(II) centres in complexes 1 and 2, confirming the presence of three and four paramagnetic, s = 1, Ni(II) centres, respectively. DFT computed magnetic exchange interactions nicely reflect experimental observations, and overlap between the magnetic orbitals can be employed to rationalise the nature and magnitude of the interactions.
Experimental section
Infra-red spectra were recorded on a Perkin Elmer FT-IR Spectrum One spectrometer equipped with a Universal ATR Sampling accessory (NUI Galway). Elemental analysis was carried out at the School of Chemistry microanalysis service at NUI Galway. Variable-temperature, solid-state direct current (dc) magnetic susceptibility data down to 5 K were collected on a Quantum Design MPMS-XL SQUID magnetometer equipped with a 7 T dc magnet. Diamagnetic corrections were applied to the observed paramagnetic susceptibilities using Pascal's constants.
Crystal structure information
Complex 3 was originally collected and published elsewhere (CCDC no.: 175223).10 The structures of 1, 2, 4 and 5 (CCDC numbers 1009473–1009476) were collected on an Xcalibur S single crystal diffractometer (Oxford Diffraction) using an enhanced Mo source. Each data reduction was carried out on the CrysAlisPro software package. The structures were solved by direct methods (SHELXS-97)16 and refined by full matrix least squares using SHELXL-97.17 SHELX operations were automated using the OSCAIL software package.18 All hydrogen atoms in 1–5 were assigned to calculated positions. All non-hydrogen atoms were refined anisotropic with the exception of the sulphate and perchlorate anions in 4 and 5 respectively, which remained isotropic. One of the two ClO4− counter anions in 5 (labelled Cl1–O50–O53) was restrained using the DFIX command. A DFIX restraint was also required for the S1–O15 bond in the SO42− anion in 4. Residual electron density in solvent accessible voids and channels were observed in 4 and so were modelled using the SQUEEZE program.19 The two large channels (total voids volume ∼1143 Å3) in 4 contained extremely diffuse electron density and were assumed to contain numerous waters of crystallisation. CHN analysis on 4 supported these observations. Although the PLATON program suggests the orthorhombic Aba2 space group for the structure in 5 and despite our best efforts, no plausible structural solution was obtained.
Computational details
DFT studies were performed on complexes 1 and 2 to predict the ground spin state of each individual Ni(II) ion and to ascertain the exchange coupling constants between ions. The calculations were performed using the Gaussian 09 suite of programmes.20 We used the hybrid B3LYP function21 along with a TZV22 basis set for Ni(II) and all other elements. Density Functional Theory along with broken symmetry23 has been shown to be a reliable tool for computing exchange coupling. For systems with two paramagnetic centres, the energy difference between the high and low spin configurations will yield a magnetic coupling constant (J). However since we are dealing with pentametallic systems the number of possible configurations is greater (2n/2; where n = number of paramagnetic centres). For complexes 1 and 2 we have used the spin Hamiltonians in eqn (1) (1) and 2 (2), which correspond to the model scheme in Fig. 10.| | |
Ĥ = −2J1A(Ŝ1·Ŝ3) − 2J1B(Ŝ1·Ŝ3A)
| (1) |
| | |
Ĥ = −2J1A(Ŝ1·Ŝ2) − 2J1B(Ŝ1·Ŝ3) − 2J2A(Ŝ2·Ŝ3) − 2J2B(Ŝ2·Ŝ5) − 2J1C(Ŝ1·Ŝ5)
| (2) |
here the Ji (i = 1–3A in 1; i = 1–5 in 2) values are the isotropic exchange coupling constants, Si the spin moment on the Ni(II) centres.
Preparation of complexes
All reactions were performed under aerobic conditions and all reagents and solvents were used as purchased. Caution: although no problems were encountered in this work, care should be taken when manipulating the potentially explosive perchlorate salts. 2-(Dimethylamino)phenylhydroxamic acid (L1H2) and 2-(amino)phenylhydroxamic acid (L2H2) were synthesised using previously reported synthetic procedures.24
[Ni5(L1)4(MeOH)4](ClO4)2·2MeOH (1). Ni(ClO4)2·6H2O (0.2 g, 0.55 mmol), L1H2 (0.1 g, 0.55 mmol) and NaOH (0.022 g, 0.55 mmol) were dissolved in 30 cm3 of MeOH and stirred for 2 h. The green solution obtained was filtered and X-ray quality crystals were obtained upon slow evaporation of the mother liquor, and from diffusion of Et2O into the mother liquor. Crystals of 1 were collected and air dried, with a yield of approximately 14%. Elemental analysis (%) calculated as [Ni5(L1)4(MeOH)2](ClO4)2·5H2O (C38H58Cl2N8O23Ni5): C 33.58, H 4.30, N 8.24. Found: C 33.36, H 4.30, N 8.24. FT-IR (cm−1): 3511(w), 1591(s), 1559(m), 1465(w), 1373(m), 1279(w), 1084(s), 1014(m), 936(m), 910(m), 777(m), 704(m), 688(m), 676(m), 663(s).
[Ni5(L1)4(py)5](ClO4)2·H2O (2). Ni(ClO4)2·6H2O (0.25 g, 0.68 mmol), L1H2 (0.12 g, 0.68 mmol) and NaOH (0.027 g, 0.68 mmol) were dissolved in 35 cm3 of MeOH. After 5 minutes 1 cm3 (12.4 mmol) of pyridine was added and the solution stirred for a further 2 h. The resultant green solution was filtered and X-ray quality crystals of 2 were obtained upon slow evaporation of the mother liquor. Crystals of 2 were also obtained by diffusing Et2O into the mother liquor. Both batches of 2 were collected and air dried with a yield of approximately 10%. Elemental analysis calculated (%) for [Ni5(L1)4(py)5](ClO4)2·3H2O (C61H71Cl2N13O19Ni5): C 44.28, H 4.33, N 11.00. Found: C 44.01, H 4.22, N 10.99. FT-IR (cm−1): 2990(w), 1590(m), 1566(w), 1541(m), 1486(w), 1467(w), 1447(m), 1375(m), 1284(w), 1218(w), 1147(w), 1082(s), 1028(m), 1014(m), 946(m), 918(m), 784(w), 765(m), 751(m), 704(m), 689(s), 673(m), 662(m).
[Ni7(L1H)8(L1)2(H2O)6](SO4)·15H2O (3). L1H2 (0.25 g, 1.4 mmol) in 20 cm3 of methanol was added to a solution of NiSO4·6H2O (0.46 g, 1.76 mmol) in 40 cm3 of water. The pH of the resulting solution was raised to 6.1 by addition of 0.2 M NaOH before being left to stand at 4 °C for 48 hours. The resulting light green semi-crystalline product was filtered and dried before recrystallisation from methanol and water (1:1). The resulting green crystalline solid of 3 was filtered, washed with methanol and air dried with a yield of approximately 40%. Elemental analysis calculated (%) for [Ni7(L1H)8(L1)2(H2O)6](SO4)·15H2O (C90H150N20O45SNi7): C 40.39, H 5.67, N 10.47. Found: C 39.98, H 5.32, N 10.19. FT-IR (cm−1): 2987(s), 2795(s) 1608(s) 1562(s), 1289(m).
[Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](SO4)·29H2O (4). Ni(SO4)·6H2O (0.25 g, 0.95 mmol), L2H2 (0.15 g, 0.95 mmol) and NEt4OH (0.7 cm3, 0.72 g, 4.89 mmol) were dissolved in 40 cm3 of a 1:1 MeOH:H2O solution. The solution was stirred for 4 h resulting in a green solution which was then filtered and allowed to stand. Upon slow evaporation green X-ray quality crystals of 4 formed after a few days. The crystals were collected and air dried with a yield of approximately 11%. Elemental analysis (%) calculated for as [Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](SO4)·12H2O (C70H96N20O40S1Ni9): C 34.77, H 4.00, N 11.59. Found: C 35.14, H 3.61, N 11.40. FT-IR (cm−1): 3200(w), 1583(m), 1547(s), 1492(m), 1450(w), 1373(m), 1152(w), 1080(m), 1017(m), 935(w), 903(m), 819(w), 747(s), 692(m), 670(s).
[Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](ClO4)2·2MeOH·18H2O (5). Ni(ClO4)2·6H2O (0.25 g, 0.68 mmol), L2H2 (0.10 g, 0.68 mmol) and NEt4OH (0.7 cm3, 0.72 g, 4.89 mmol) were dissolved in 40 cm3 of a 1:1 MeOH:CH3CN solution. The solution was stirred for 4 h resulting in a green solution which was filtered and evaporated to dryness. The green solid was subsequently re-dissolved in 20 cm3 of a 1:1 MeOH:H2O solution and stirred for a further 2 h. The resultant green solution was filtered and X-ray quality crystals of 5 were obtained upon slow evaporation of the reaction mixture in 10% yield. Elemental analysis calculated (%) for [Ni9(μ-H2O)2(L2)6(L2H)4(H2O)2](ClO4)2·20H2O (C70H112N20O52Cl2Ni9): C 31.55, H 4.24, N 10.51. Found: C 31.82, H 3.92, N 10.25. FT-IR (cm−1): 3203(m), 1611(m), 1583(m), 1547(s), 1494(m), 1450(w), 1374(m), 1153(m), 1091(m), 1014(m), 936(m), 903(s), 869(w), 819(w), 749(s), 694(m), 671(s).
Acknowledgements
The authors would like to thank the College of Science, NUI Galway for their support (CM). EKB would like to thank the EPSRC for funding (SS). GR acknowledges financial support from the Government of India through the Department of Science and Technology (SR/S1/IC-41/2010; SR/NM/NS-1119/2011) and generous computational resources from the Indian Institute of Technology-Bombay. MKS thanks the University Grants Commission for a Junior Research Fellowship.
References
-
(a) D. Griffith, M. Devocelle and C. J. Marmion. Hydroxamic Acids: Their Chemistry, Bioactivity, Solution and Solid Phase Synthesis in Amino Acids, Peptides and Proteins in Organic Chemistry, ed. A. B. Hughes, 2009, vol. 2, pp. 93–137 Search PubMed;
(b) M. Paris, M. Porcelloni, M. Binaschi and D. Fattori, J. Med. Chem., 2008, 51, 1505 CrossRef CAS PubMed;
(c) Z. Amtul, R. Attaur, R. A. Siddiqui and M. I. Choudhary, Curr. Med. Chem., 2002, 9, 1323–1348 CrossRef CAS;
(d) D. T. Puerta and S. M. Cohen, Curr. Top. Med. Chem., 2004, 4, 1551 CrossRef CAS.
-
(a) E. M. F. Muri, M. J. Nieto, R. D. Sindelar and J. S. Williamson, Curr. Med. Chem., 2002, 9, 1631–1653 CrossRef CAS;
(b) C. J. Marmion, D. Griffith and K. B. Nolan, Eur. J. Inorg. Chem., 2004, 2004, 3003 CrossRef PubMed.
- R. Codd, Coord. Chem. Rev., 2008, 252, 1387–1408 CrossRef CAS PubMed.
-
(a) A. M. Wilson, P. J. Bailey, P. A. Tasker, J. R. Turkington, R. A. Grant and J. B. Love, Chem. Soc. Rev., 2014, 43, 123–134 RSC;
(b) B. K. Tait, K. E. Mdlalose and I. Taljaard, Hydrometallurgy, 1995, 38, 1–6 CrossRef CAS.
- For an extensive review on metallacrowns see: G. Mezei, C. M. Zaleski and V. L. Pecoraro, Chem. Rev., 2007, 107, 4933 CrossRef CAS PubMed.
- C. McDonald, T. Whyte, S. M. Taylor, S. Sanz, E. K. Brechin, D. Gaynor and L. F. Jones, CrystEngComm, 2013, 15(34), 6672–6681 RSC.
- A. W. Addison, T. N. Rao, J. Reedjik, J. van Rijn and G. C. Verschoor, J. Chem. Soc., Dalton Trans., 1984, 1349–1356 RSC.
-
(a) Z. Chen, M. Jia, Z. Zhang and F. Liang, Cryst. Growth Des., 2010, 10(11), 4806–4814 CrossRef CAS;
(b) M. Alexiou, I. Tsivikas, C. Dendrinou-Samara, A. A. Pantazaki, P. Trikalitis, N. Lalioti, D. A. Kyriakidis and D. P. Kessissogolou, J. Inorg. Biochem., 2003, 93, 256–264 CrossRef CAS;
(c) G. Psomas, A. J. Stemmler, C. Dendrinou-Samara, J. J. Bodwin, M. Schneider, M. Alexiou, J. W. Kampf, D. P. Kessissoglou and V. L. Pecoraro, Inorg. Chem., 2001, 40, 1562–1570 CrossRef CAS PubMed;
(d) D. Bacco, V. Bertolasi, F. Dellavalle, L. Galliera, N. Marchetti, L. Marchio, M. Remelli and M. Tegoni, Dalton Trans., 2011, 40, 2491–2501 RSC.
- G. Psomas, C. Dendrinou-Samara, M. Alexiou, A. Tsohos, C. P. Raptopoulou, A. Terzis and D. P. Kessissoglou, Inorg. Chem., 1998, 37, 6556–6557 CrossRef CAS.
- D. Gaynor, Z. A. Starikova, S. Ostrovsky, W. Haase and K. B. Nolan, Chem. Commun., 2002, 506 RSC.
- For examples of diamagnetic square based pyramidal Ni(II) complexes see:
(a) A. F. Kolodziej, Prog. Inorg. Chem., 1994, 41, 493 CrossRef CAS PubMed;
(b) M. A. Halcrow and G. Christou, Chem. Rev., 1994, 94, 2421 CrossRef CAS;
(c) J. J. Wilker, A. Gelasco, M. A. Pressler, R. O. Day and M. J. Maroney, J. Am. Chem. Soc., 1991, 113, 6324 CrossRef;
(d) M. Cha, C. L. Gatlin, S. C. Critchlow and J. A. Kovacs, Inorg. Chem., 1993, 32, 5868 CrossRef CAS;
(e) D. C. Goodman, P. J. Farmer, M. Y. Darensbourg and J. H. Reibenspies, Inorg. Chem., 1996, 35, 4989 CrossRef CAS PubMed and references therein. For examples of paramagnetic square pyramidal Ni(II) complexes see;
(f) S. Brooker and P. D. Croucher, Chem. Commun., 1997, 459 RSC;
(g) U. Lehmann, J. Lach, C. Loose, T. Hahn, B. Kersting and J. Kortus, Dalton Trans., 2013, 42, 987 RSC.
- W. H. Press, S. A. Teukolsky, W. T. Vetterling and B. P. Flannery, Numerical Recipes in C: The Art of Scientific Computing, Cambridge University Press, Cambridge, 2nd edn, 1992 Search PubMed.
-
(a) K. K. Nanda, L. K. Thomson, J. N. Bridson and K. Nag, J. Chem. Soc., Chem. Commun., 1994, 1337–1338 RSC;
(b) K. K. Nanda, R. Das, L. K. Thomson, K. Venkatsubramanian, P. Paul and K. Nag, Inorg. Chem., 1994, 33, 1188–1193 CrossRef CAS;
(c) K. K. Nanda, R. Das, L. K. Thomson, K. Venkatsubramanian and K. Nag, Inorg. Chem., 1994, 33, 5934–5939 CrossRef CAS;
(d) A. J. Stemmler, J. W. Kampf, M. L. Kirk and V. L. Pecoraro, J. Am. Chem. Soc., 1995, 117, 6368–6369 CrossRef CAS;
(e) D. Arnold, D. A. Brown, O. Deeg, W. Errington, W. Haase, K. Herlihy, T. J. Kemp, H. Nimir and R. Werner, Inorg. Chem., 1998, 37, 2920–2925 CrossRef.
- B. Biswas, U. Pieper, T. Weyhermuller and P. Chaudhuri, Inorg. Chem., 2009, 48, 6781–6793 CrossRef CAS PubMed.
- A. Upadhyay, J. Rajpurohit, M. K. Singh, R. Dubey, A. K. Srivastava, A. Kumar, G. Rajaraman and M. Shanmugam, Chem.–Eur. J., 2014, 20, 6061–6070 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 467 CrossRef.
- G. M. Sheldrick, SHELXL-97, A computer programme for crystal structure determination, University of Gottingen, 1997 Search PubMed.
- P. McArdle, P. Daly and D. Cunningham, J. Appl. Crystallogr., 2002, 35, 378 CrossRef CAS.
-
(a) A. Spek, J. Appl. Crystallogr., 2003, 36, 7–13 CrossRef CAS;
(b) P. van der Sluis and A. L. Spek, Acta Crystallogr., Sect. A: Found. Crystallogr., 1990, 46, 194–201 CrossRef.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. Austin, J. R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09. 2009 Search PubMed.
-
(a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS PubMed;
(b) A. D. Becke, J. Chem. Phys., 1993, 98, 1372 CrossRef CAS PubMed;
(c) C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter, 1988, 37, 785 CrossRef CAS;
(d) P. J. Stephens, F. J. Devlin, C. F. Chabalowski and M. J. Frisch, J. Phys. Chem., 1994, 98, 11623 CrossRef CAS.
-
(a) A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS PubMed;
(b) A. Schaefer, C. Huber and R. Ahlrichs, J. Chem. Phys., 1994, 100, 5829 CrossRef CAS PubMed;
(c) G. E. Scuseria and H. F. Schaefer III, J. Chem. Phys., 1989, 90, 3700 CrossRef CAS PubMed.
- J. J. Borras-Almenar, J. M. Clemente-Juan, E. Coronado and B. S. Tsukerblat, J. Comput. Chem., 2001, 22, 985 CrossRef CAS PubMed.
- D. Gaynor, Z. A. Starikova, W. Haase and K. B. Nolan, Dalton Trans., 2001, 1578 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1009473–1009476. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra06064d |
| ‡ Current address: School of Chemistry, Bangor University, Bangor, Gwynedd, Wales, LL57 2DG, UK. E-mail: E-mail: leigh.jones@bangor.ac.uk; Tel: +44 (0)1248-38-2391. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here.