
A ratiometric fluorescent probe for fluoride ion based on naphthoimidazolium receptor†
Abstract
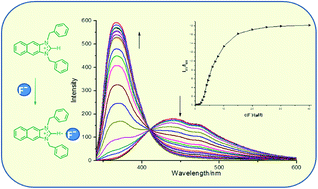
Three imidazolium derivatives 3–5 were designed and synthesized, in which naphthaimidazolium group acted as both fluorophore and anion receptor. Compound 3 exhibited high selectivity for F− in CH3CN solution over all the other anions and acted as a ratiometric fluorescent probe for F− with an enhanced blue-shift in emission. However, the fluorescence of compound 4 and 5 displayed a quenched blue-shift in emission with fluoride ion and could be quenched by some other tested anions, where the degree of quenching depended on the characteristic of the anions. More importantly, only compound 3 could detect F− in DMSO–water (95 : 5, v/v) aqueous solution ratiometrically. Based on the analysis of the results of 1H-NMR and 19F-NMR, it was deduced that compound 3 bound with F− mainly by the force of hydrogen bonding, while compound 4 and 5 coordinated with F− through electrostatic interaction.
 Please wait while we load your content...
Please wait while we load your content...

