
Photophysical, bandstructural, and textural properties of o-FeNbO4 in relation to its cocatalyst-assisted photoactivity for water oxidation
Robin Babua,
Sarika Kelkarb,
Vikas Kashidc,
Srungarpu N. Acharyd,
Hemant G. Salunkee and
Narendra M. Gupta*a
aCatalysis Division, National Chemical Laboratory, Dr Homi Bhabha Road, Pune 411008, India. E-mail: nm.gupta@ncl.res.in; Fax: +91-22-25902633; Tel: +91-20-25902008
bPhysical and Materials Chemistry Division, National Chemical Laboratory, Dr Homi Bhabha Road, Pune 411008, India
cDepartment of Physics, University of Pune, Pune 411007, India
dChemistry Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
eTechnical Physics Division, Bhabha Atomic Research Centre, Trombay, Mumbai 400085, India
First published on 21st July 2014
Abstract
In this study, a relationship between physicochemical, photophysical and photocatalytic properties of hydrothermally synthesized orthorhombic iron niobate (FeNbO4) is investigated. o-FeNbO4 displayed a multi-regime optical absorbance, which was ascribed to at least two distinct phenomena: (i) bandgap (∼3.4 eV) excitation giving rise to UV absorbance and (ii) energy transitions involving disorder-induced sub-bandgap donor or acceptor states leading to visible light absorbance. The preparation-dependent distortion in the crystal lattice and the existence of closely spaced inter-bandgap energy states were corroborated by powder X-ray diffraction, photoluminescence, thermoluminescence, and Raman spectroscopy studies. The first principles electronic structure elucidation and photoelectrochemical measurements supported a wide bandgap for FeNbO4, in contrast to the narrow bandgap reported previously. Correspondingly, a small photocurrent density was observed for FeNbO4 (∼2 to 3 μA cm−2) under 1 sun illumination, suggesting the availability of a smaller cross section of photogenerated charge pairs. Following these band characteristics, while no H2 evolution was observed, FeNbO4 gave rise to particle size-dependent O2 evolution during visible light irradiation of water in the presence of electron scavengers, the samples loaded with NiO as cocatalyst showing better activity. Further, the transmission electron microscopy examination revealed the dominant exposure of (011) facets of FeNbO4, besides a significant heterogeneity of inter-domain boundaries. Overall, our results confirm that the photoactivity of metal/oxide nanocomposites is governed by a combination of factors, such as: grain morphology, microstructure, surface adsorption states, and the localized inter-bandgap energy states. Our study also reveals that, in contrast to prevalent assumptions, the wavelength at the absorption edge may not represent the true band-to-band energy gap of metal oxide semiconductors, which is relevant to their photocatalytic activity.
1. Introduction
Despite extensive research efforts devoted already to semiconductor-mediated photocatalytic splitting of water, the efficient and viable production of hydrogen fuel continues to elude us. The lack of sustainable photocatalysts with requisite band structures and quantum efficiencies is primarily responsible for this situation. Moreover, a clear understanding of the physicochemical and photophysical properties governing the photoactivities of semiconducting materials remains to be achieved. Among the numerous metal oxide/sulfide metal composites that have been investigated, certain metal niobate photocatalysts have drawn considerable interest for such applications.1–5 The merit of metal niobates as photocatalysts lies in their visible-light sensitivity, arising from distorted octahedral [NbO6] units, along with the fairly high energy level of the Nb 4d orbital. In recent studies on the photocatalytic properties of metal niobates, we demonstrated how impurity-induced lattice imperfections, grain morphologies, and metal-semiconductor hetero-junctions may play a crucial role in the water splitting activity of gallium niobate.6 In the same context, we envisioned that iron niobate (FeNbO4), a scantily investigated compound, may serve as a potential visible light water splitting photocatalyst, owing to its reported bandgap energy of ∼2.0 eV, optimum flat band potential at −0.4 V (SCE), and its significant activity for the visible light degradation of dyes.7,8Nano-structured photocatalysts were synthesized in our study by hydrothermal method, thus enabling the preparation of pure, orthorhombic phase FeNbO4 samples at a temperature as low as 600 °C, compared to the conventional temperature of >1100 °C by solid-state procedures.7,9 Post-synthesis calcination at two different temperatures, 600 or 800 °C, allowed for the preparation of a set of samples comprising particles of two different size ranges. The synthesized samples were coated with a small amount of Pt or NiO as a co-catalyst. Powder X-ray diffraction (XRD), diffuse reflectance UV-visible (DR UV-Vis) absorption, photoluminescence (PL), thermoluminescence (TL), electron microscopy (EM), and laser-Raman spectroscopy were employed to systematically characterize the samples. The photoactivity of FeNbO4 was tested for the dissociation of water under visible light irradiation and in the presence of several well known electron donor and electron acceptor sacrificial reagents. The emphasis in this study was on investigating the crystallographic, morphological, optical and microstructural properties responsible for the photoactivity of metal/oxide nanocomposites, using Pt/FeNbO4 and NiO/FeNbO4 as model photocatalysts. The first-principles-based density functional theory calculations were performed with a view to discern the band-structural features that may account for photophysical and photocatalytic properties of the studied materials.
2. Experimental section
2.1 Sample preparation
For the hydrothermal preparation of a representative batch of ∼1 g sample, 7.5 mmol of Fe(NO3)3·9H2O (≥98% purity, Sigma-Aldrich) was dissolved in 15 mL distilled water with constant stirring. Similarly, 7.5 mmol of NbCl5 (≥99.9% purity, Sigma-Aldrich) was dissolved in 15 mL absolute ethanol (99.9% purity, Cympran Gludt BV, Belgium). These two solutions were mixed via stirring and the pH was adjusted to ∼10 by addition of ammonia (25%) solution, followed by further stirring. The mixture was heated (∼180 °C, 24 h) in a sealed, Teflon-lined autoclave, followed by slow cooling to room temperature. The reddish brown precipitate obtained on centrifugation was washed repeatedly with distilled water and then ethanol. After drying at 80 °C in an air oven, the dry gel was powdered and sieved to <60 μm grain size. The sample was then calcined in air, either at 600 or 800 °C (8 h, 5 °C min−1) and ground again. The FeNbO4 samples calcined at 600 and 800 °C are designated as FN-6 and FN-8, respectively, for brevity.Pt and NiO-loaded photocatalysts were prepared by the incipient wet impregnation of an aliquot of FeNbO4 powder with a requisite amount of either hexachloroplatinic acid (H2PtCl6·2H2O) or nickel nitrate (Ni(NO3)2·6H2O, Sigma Aldrich) with continuous stirring. The mass was dried slowly over a water bath and the sample was then subjected to heat treatment at 400 °C for 4 h. The dispersed platinum was then converted into its metallic state by exposure to UV radiation (300 W mercury vapour lamp) for ∼12 h. No such treatment was applied to the Ni-coated samples. The oxidation states of Pt0 and Ni2+ and the co-catalyst content in the photocatalysts were determined by X-ray photoelectron spectroscopy (XPS) and energy dispersive X-ray analysis EDX.
2.2 Characterization
Following instrumentation were adopted for sample characterization: powder XRD patterns (PanAnalytical diffractometer, CuKα radiation, scan rate 1° min−1), DR UV-Vis spectra (Perkin Elmer Lambda-650 spectrophotometer, BaSO4 as reference), PL spectra (Horiba Scientific Fluoromax-4 spectrofluorimeter, λexc = 420 nm), laser Raman spectra (Horiba JY Lab RAM HR800 spectrometer, 633 nm laser for excitation, spectral resolution of 0.35 cm−1). The grain morphology was examined on scanning electron microscope (SEM; FEI company, model-Quanta 200 3D, tungsten filament, acceleration voltage 20 kV). The microstructure of the samples was examined from high-resolution transmission electron microscopy (HR-TEM) images (Tecnai model-GF 30 microscope, FEI Company, Netherlands, 300 kV field emission gun). The N2-adsorption BET specific surface area (SA) was measured on a Quadrasob-SI. The samples were evacuated at 250 °C prior to SA measurements.2.3 Activity measurements
The photoactivity was monitored in a closed quartz photo reactor, positioned at ∼6 cm from the light source. Two alternative illumination sources were employed including a 1 sun Newport solar simulator (500 W Xe arc lamp, irradiance 50 × 105 Lx) or a 300 W Xe lamp (M. Watanabe & Co, Japan, model XDS-301S, irradiance 1 × 105 Lx). These experiments were conducted with or without a UV cut-off filter with transmission at >395 nm (CVI Meller Griot CG-GG-395-50) in the path of the light beam. Various concentrations of electron donor or electron acceptor sacrificial reagents such as CH3OH, Na2CO3, KOH, and AgNO3 were investigated in the photoactivity measurements. In a typical experiment using silver nitrate as an electron scavenger, 0.06 g of the sample powder was suspended in 15 mL of aqueous AgNO3 (10 mM) solution. The reaction products were analysed at regular intervals over a 5–6 h period using a gas chromatograph (Agilent model-India, packed Molecular Sieve 5A column, thermal conductivity detector maintained at 300 K, argon as carrier gas).2.4 Band characteristics
The band gap of the photocatalysts was estimated from optical absorption spectra by extrapolation of the linear region in Tauc equation plots: (αhν)2 vs. (hν) for a direct band gap semiconductor and (αhν)1/2 vs. (hν) for an indirect band gap material, where α is the absorption coefficient and hν is photon energy. The valence band (VB) and conduction band (CB) potentials were estimated from the absolute electronegativity of the atoms using an empirical expression:| Evb = χ − E0 + 1/2Eg, | (1) |
Mott–Schottky (M–S) plots were recorded to estimate the electrochemical flat band potential of FeNbO4 with respect to the redox potential of water. For these measurements, a film of FeNbO4 was deposited onto a fluorine-doped, titanium dioxide-coated glass plate by the doctor blade method. A three-electrode measurement setup (Autolab PGSTAT 30-Eco-Chemie potentiostat) was utilized for recording the M–S plots in the dark, using an AC impedance measurement program with 10 mV ac amplitude and 10 kHz frequency. In these experiments, the sample film served as the working electrode, Pt foil as the counter electrode, and saturated calomel as the reference electrode. The electrolyte used was 1 M KOH (pH = 13.8). The M–S plots were obtained by plotting C−2 against the applied voltage (Edc), where C is the observed capacitance. The current vs. voltage (I–V) plots were recorded during illumination with a 1 sun Newport solar simulator equipped with a chopping light facility to achieve alternating dark and light conditions.
2.5 Thermoluminescence (TL) study
To record the TL glow curves, catalyst samples were exposed to UV irradiation for 1 h (6 W GT5 lamps, λmax ∼ 254 nm). After ∼10 min cooling time, TL readouts were recorded at a heating rate of ∼1 °C s−1 on a TL reader Risø TL/OSL-DA-20 (Risø National Laboratory, Denmark, Bialkali EMI 9235 QB photomultiplier tube). An optical filter was interposed between the photomultiplier tube and sample holder to cut off infrared radiation. Experiments were also conducted on samples exposed to white light radiation from a 200 W tungsten lamp.2.6 Electronic structure
Ab initio first principles spin polarised electronic structure calculations for orthorhombic structure of FeNbO4 (space group: Pbcn, space group no. 60) were conducted using a Vienna Ab initio Simulations Package (VASP).10 In this structure Fe and Nb occupy 4c crystallographic positions with 50% occupancy, while O occupies 8d positions. The Projector Augmented Wave method (PAW) was used for the description of electron–ion interactions and the plane waves were included up to high energetic cut-off of 400 eV. The Brillouin zone was integrated using a converged 15 × 15 × 15 Monkhorst–Pack grid of k-points. This gave approximately 2330 number of plane waves per k-point in the Brillouin zone. The self consistency loops were repeated until the energy difference became less than 10−5 eV. The FeNbO4 unit cell was optimized for the reported lattice constant values of: a = 4.627 Å, b = 5.5898 Å and c = 4.9741 Å. The optimization was performed using LSDA + U, with effective on-site Coulomb interaction parameters (Ueff) for Fe and Nb equal to 10 eV and 8 eV, respectively, where the effective on-site exchange interaction parameters (Jeff) for Fe and Nb equal to 1 eV and 0.8 eV, respectively.3. Results
3.1 Morphology and microstructure
As shown in the SEM images in Fig. 1, FN-6 (a) and FN-8 (b) samples were comprised of irregular shape granules having a cross-sectional size of 40–80 nm and 75–120 nm, respectively. The TEM images (Fig. 1c and d) revealed a spherical shape for primary particles, the size of the particles being larger for a high temperature calcined sample FN-8 (Fig. 1d) compared to that for FN-6 (Fig. 1c). Also, the sizes of the primary particles were marginally larger for metal-coated samples. The particle size ranges in different samples, estimated from ca. 50 to 70 particles in multiple TEM images for an individual sample, are compiled in Table 1. The high-resolution TEM images revealed a significant variation in crystalline nature and microstructure of the samples calcined at the two different temperatures. Thus, the HRTEM images of FN-6 comprised of nanodomains of varying size, both without and with presence of lattice fringes. This indicated a rather poor crystalline nature of FN-6. On the other hand, the HRTEM images of FN-8 exhibited randomly distributed domains of long-range lattice fringes. The panels (e) and (f) in Fig. 1 respectively show a representative HRTEM image of FN-8 and a corresponding live FFT picture. A remarkable feature of these images is the dominant exposure of (011) facets, against the most intense (I100) lattice plane (111) of o-FeNbO4 (PCPDF: #84-1981). As reported,11 the photoactivity of a metal oxide, such as TiO2, is controlled by the nature of an exposed lattice plane, because of its unique geometrical and electronic structure. A significant heterogeneity of the inter-domain contacts, which in turn may lead to distinct surface states, is another noticeable feature of Fig. 1e. It needs a mention that the HRTEM images recorded on Pt/FN-8 and Pt/FN-6 samples (not shown) revealed a highly dispersed state of the cocatalyst. The Pt particles were normally of spherical shape, with their cross sectional size varying from ∼4 to 10 nm.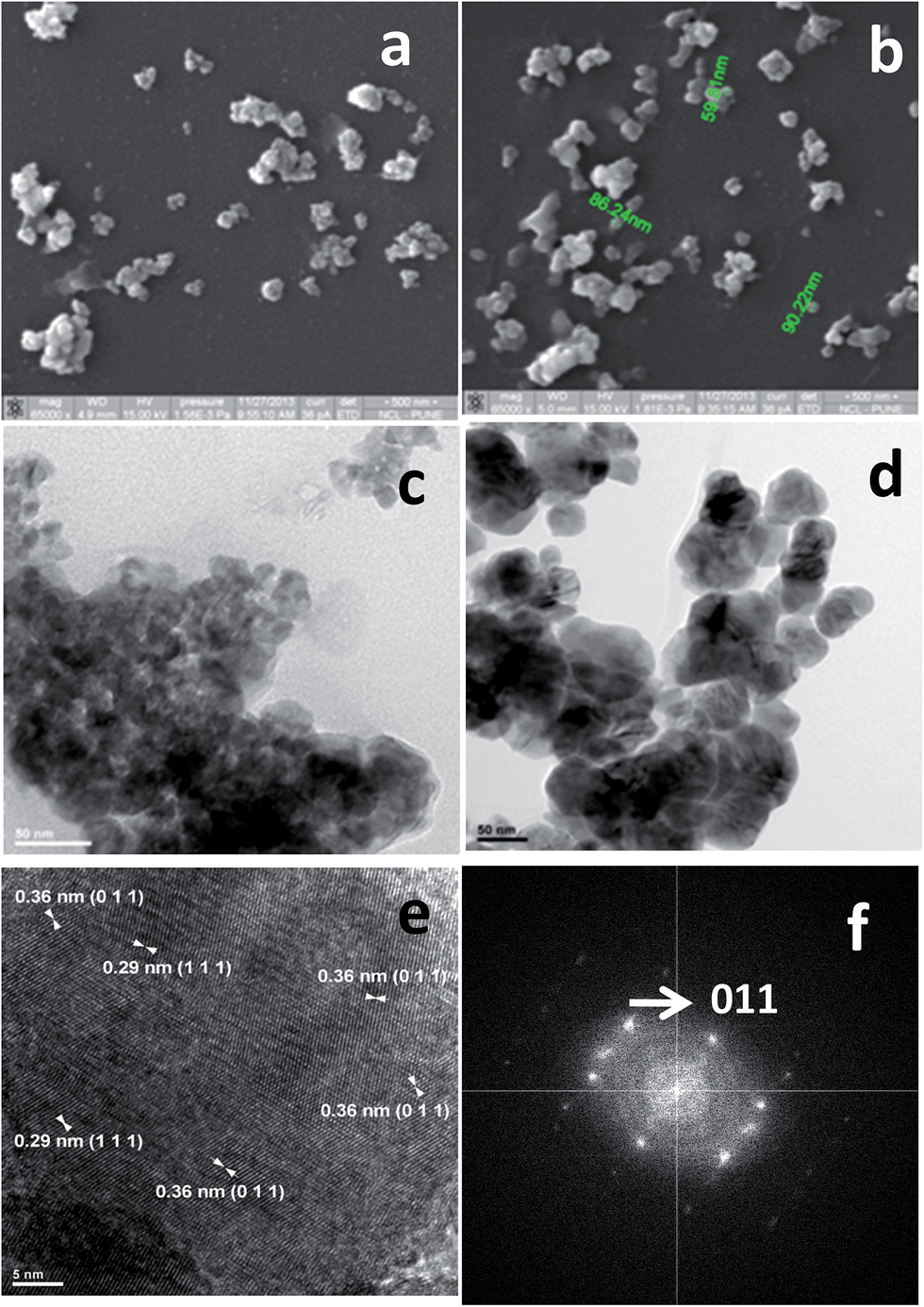 | ||
| Fig. 1 Scanning electron micrographs (a and b) and transmission electron micrographs (c and d) of FN-6 (a and c) and FN-8 (b and d) photocatalysts. The panels (e and f) show a representative high resolution TEM image of FN-8 and a corresponding live FFT image, respectively. | ||
| S.N | Sample | Preparation details | Surface areaa ± 10% m2 g−1 | Average particle sizeb nm | Primary particle sizec nm |
|---|---|---|---|---|---|
| a From N2 adsorption at liquid nitrogen temperature.b From XRD line width.c From TEM images. | |||||
| 1 | FN-6 | Hydrothermal, calcination at 600 °C | 45.2 | 9.7 | 8–16 |
| 2 | FN-8 | Hydrothermal, calcination at 800 °C | 19.8 | 25.6 | 20–45 |
| 3 | Pt/FN-6 | 1 wt% Pt dispersed over FN-6 | 34.6 | 15.7 | 12–35 |
| 4 | Pt/FN-8 | 1 wt% Pt dispersed over FN-8 | 13.2 | 26.9 | 25–55.6 |
| 5 | NiO-1/FN-6 | 1% NiO dispersed over FN-6 | 49.5 | 7.8 | — |
| 6 | NiO-2/FN-6 | 2% NiO dispersed over FN-6 | 56.7 | 6.4 | — |
3.2 X-ray diffraction studies
Fig. 2 shows the XRD patterns of an un-calcined sample (curve c), and for the samples calcined at 600 °C (FN-6, curve d) or 800 °C (FN-8, curve f). The XRD patterns of Pt- and NiO-coated FeNbO4 were similar to those shown in Fig. 2, with the exception of a minor difference in the line widths. Curve (e) depicts a typical XRD pattern of NiO-2/FN-6. The vertical lines in traces (a) and (b) of Fig. 2 represent the peak positions reported for NiO (JCPDS-PDF #40-1049) and orthorhombic FeNbO4 (JCPDS-PDF #84-1981), respectively. Comparatively broader XRD peaks in Fig. 2d reveal the poor crystalline nature of FN-6, due to the presence of smaller particles in the sample, in harmony with the microscopy results described above. The average sizes of the crystallites in different photocatalyst samples, determined by applying the Scherrer equation to the full width at half maximum of the prominent XRD lines, are included in Table 1. The absence of clearly discernible XRD reflections of NiO in Fig. 2e (cf. Fig. 1a) is accountable to a highly dispersed state of the cocatalyst, in tune with our HRTEM results. We may mention that the above-mentioned calcination-dependent particle size data obtained from XRD line widths find a good match in the specific surface area of different FeNbO4 photocatalysts, as determined from BET (N2 adsorption) measurements and compiled in Table 1. | ||
| Fig. 2 Powder X-ray diffraction patterns of FeNbO4 photocatalysts: (c) un-calcined sample, (d) FN-6, (e) NiO-2/FN-6, and (f) FN-8 (without and with Rietveld refinement). Plots (a) and (b) show PCPDF patterns for NiO (JCPDS-PDF #40-1049) and orthorhombic FeNbO4 (JCPDS-PDF #84-1981), respectively. Plot (g) is difference of the observed and calculated XRD pattern of FN-8. Topmost vertical ticks indicate Bragg positions for o-FeNbO4. | ||
XRD patterns in Fig. 2d and f match perfectly with the α-PbO2 type cation-disordered structure (Pbcn) of o-FeNbO4 with the metal ions coordinated octahedrally with oxygen ions.12,13 As reported,12,13 the cation ordering and distortion in FeO6 or NbO6 octahedra are known to depend on the polymorphic structure of FeNbO4. Among the three known polymorphs, the orthorhombic and tetragonal rutile (P42/mnm)-type FeNbO4 has a cation disordered structure, while the monoclinic phase possesses a cation ordered structure.13 The XRD patterns in Fig. 2d and f thus reveal that the hydrothermal preparation of pure orthorhombic phase FeNbO4 leads to a cation disordered amorphous phase, the onset of which occurs at a temperature as low as 600 °C. This disorder is retained in the crystalline form obtained at 800 °C (Fig. 2f).
Fig. 3 exhibits the projected crystal structure of FeNbO4, where the MO6 (M = Fe3+, Nb5+) octahedra are linked by sharing two edges and forming zig-zag chains along the c-axis. These octahedral chains are connected by sharing the corner oxygen atoms along the a- and b-axes. Thus, the oxygen atoms have a triangular coordination with the metal ions. This octahedral arrangement is similar to that in the rutile structure, except the linear chain of the rutile structure appeared as zig-zag chains in the o-FeNbO4 structure.13,14 To determine the lattice parameters, the powder XRD pattern of a crystalline FeNbO4 sample (FN-8) was refined by the Rietveld method using GSAS software packages.15 The background of the XRD data was modelled by linear interpolation of the selected varying points. The Bragg peaks were calculated using the pseudo-Voigt function. No preferred orientation function was used for the refinement. The powder XRD pattern of FN-8 could be fitted with the structural parameters reported for o-FeNbO4, as shown in Fig. 2f. A difference plot obtained from the observed and calculated XRD patterns of FN-8 is shown in curve (g), confirming a precise fit. The calculated values for the unit cell and other structural parameters of FN-8 are compiled in Table 2.
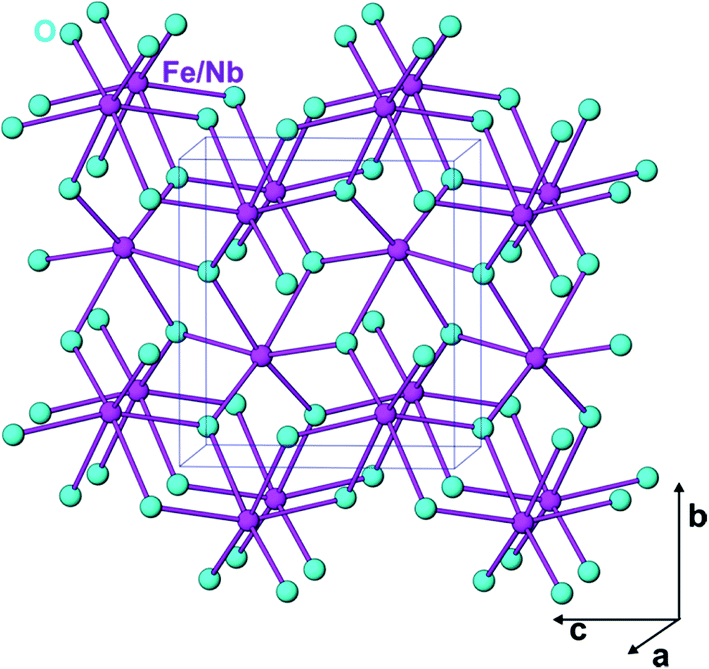 | ||
| Fig. 3 Projected crystal structure of o-FeNbO4 depicting distinct Fe(Nb)–O bond lengths. | ||
| Wyc. | x | y | z | Biso (Å)2 | Occ. | |
|---|---|---|---|---|---|---|
| a Orthorhombic (Pbcn, no. 60) a = 4.6270(4) Å; b = 5.5898(4) Å; c = 4.9741(4) Å, V = 128.65(2) Å3; Z = 2, Rp: 1.0%, Rwp: 1.6%, RB = 3.63%, RF = 2.81 (Fe/Nb)–O = 1.882(3) × 2; (Fe/Nb)–O = 2.098(4) × 2; (Fe/Nb)–O = 2.045(4) × 2, octahedral distortion: 20.9 × 10−4. | ||||||
| Fe | 4c | 0 | 0.1706(2) | 1/4 | 2.48(3) | 0.5 |
| Nb | 4c | 0 | 0.1706(2) | 1/4 | 2.48(3) | 0.5 |
| O | 8d | 0.2593(7) | 0.3867(7) | 0.0887(8) | 1.71(9) | 1.0 |
3.3 Laser Raman spectra
Fig. 4 exhibits the room temperature laser Raman spectra of FN-6 and FN-8 photocatalysts and corresponding Pt-dispersed samples. Considering the Fe/Nb as a single site in Pbcn symmetry, group theory predicts 16 Raman active modes for FeNbO4. At least 15 distinct vibrational bands are identifiable in the laser Raman spectrum of crystalline FeNbO4 (FN-8), and the prominent modes are at 816, 604, 496, 363, 306, 273, 226, 150, and 137 cm−1 (Fig. 4a). The observed Raman modes are in good agreement with a previously reported Raman spectrum of FeNbO4.16 Considerable overlap and a broadening of these bands in the Raman spectrum of the low-temperature calcined sample FN-6 (Fig. 4b) is noticeable. Similarly, a significant broadening was observed in the laser Raman spectra of Pt-loaded photocatalysts (Fig. 4c and d). Based on the previously described assignments of the niobate structure, the Raman peaks in the 800–300 cm−1 frequency region are associated with the internal vibrational modes of NbO6 octahedra, whereas the frequencies below 300 cm−1 originate from external lattice vibrations.17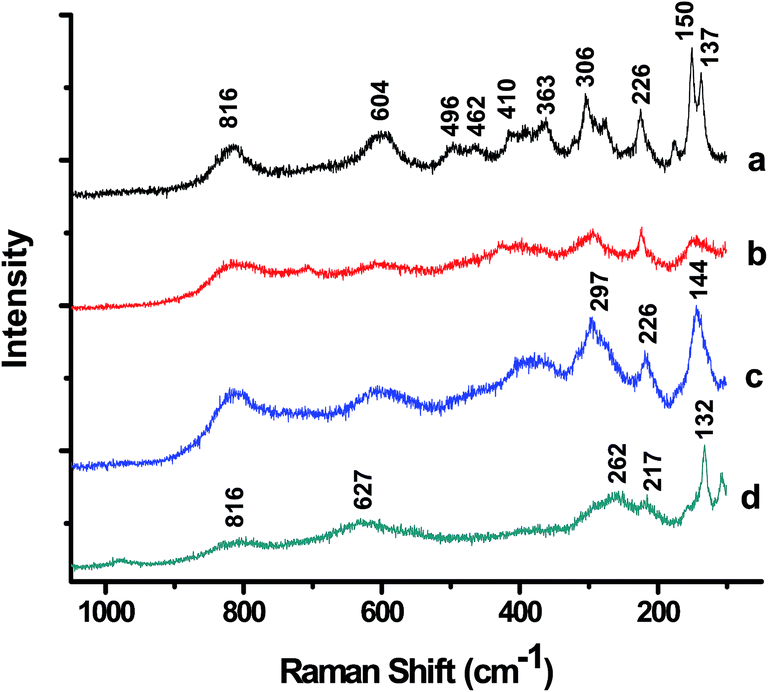 | ||
| Fig. 4 Laser Raman spectra of (a) FN-8, (b) FN-6, (c) Pt/FN-8, and (d) Pt/FN-6 photocatalysts. | ||
3.4 Optical absorbance
Fig. 5 presents the typical DR UV-visible absorbance spectra of FeNbO4 photocatalysts, without and with dispersed co-catalysts. At least three strongly overlapping absorption bands are observed in each spectrum with peak maxima at around 300, 380, and 510 nm. An enhanced absorbance in the visible region (600–700 nm) in NiO- and Pt-loaded samples (curves c and d) is characteristic of the plasmonic absorption associated with nano-sized metal particles. The Tauc equation plots between (αhν)2 versus hν gave a better fit for the absorbance data (Fig. 5 inset), suggesting a direct band energy gap of FeNbO4. The plot in the inset reveals at least three distinct energy gaps for crystalline FeNbO4, namely 2.0, 2.4, and 3.4 eV. The position of the conduction band potential (Ec) vs. NHE, deduced using eqn (1) is estimated to be +0.52, +0.32, and −0.18 eV against the valence band to conduction band energy gap values of 2.0, 2.4, and 3.4 eV, respectively. The representative band characteristics of FN-8, based on the UV-Vis results of Fig. 5, are compiled in Table 3.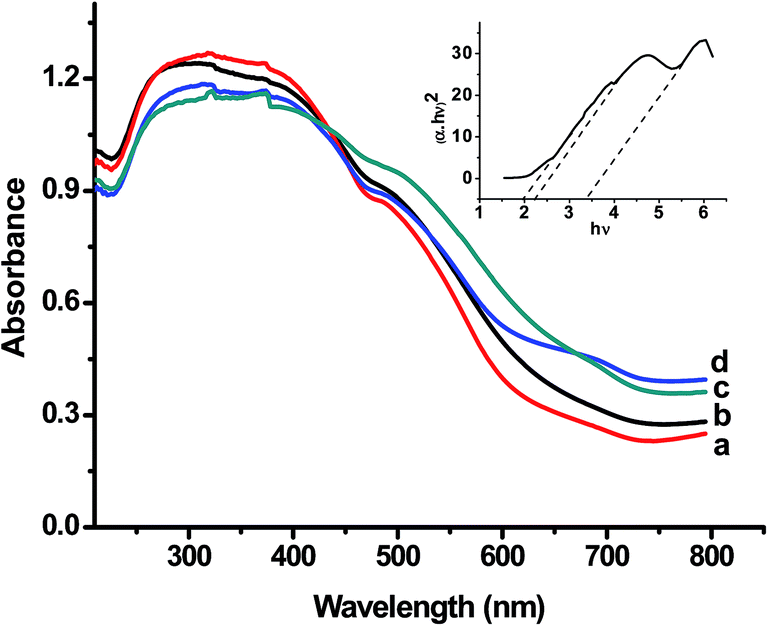 | ||
| Fig. 5 Diffuse reflectance UV-Vis absorbance spectra of FN-8 (curve a), FN-6 (curve b), NiO-1/FN-6 (curve c), and Pt/FN-8 (curve d) photocatalysts. Inset shows the Tauc equation plot (direct band gap) for FN-8. | ||
| Band gap, Eg, eV | Conduction band potential (Ecb), eV | Valence band potential (Evb), eV |
|---|---|---|
| 2.0 | +0.52 | 2.52 |
| 2.4 | +0.32 | 2.72 |
| 3.4 | −0.18 | 3.22 |
3.5 Photoluminescence (PL) studies
Fig. 6 shows representative PL spectra of different photocatalysts without (curves a and b) and with Pt-loading (curves c and d). Two intense emission bands at ∼570 and 690 nm can be observed in addition to several weak bands. The deep-level PL bands, appearing at wavelengths above the edge position, are known to arise from certain lattice defects in wide gap metal oxides, such as oxygen ion vacancies and interstitial oxygen ions.18 Furthermore, the PL quenching in the presence of dispersed Pt particles, as revealed in Fig. 6c and d, is in accord with the widely reported charge transfer effects in metal/semiconductor composite photocatalysts.19 Also, the luminescence emission in the visible region relates to the structural defects in wide gap metal oxides.20,21 Accordingly, the overlapping luminescence emission bands in the 550–700 nm range can be ascribed to discreet charge trapping states in the energy range of 1.7 to 2.2 eV; the radiative recombination of photoexcited e−/h+ pair give rise to visible light emission (Fig. 6). The existence of such inter-bandgap energy states in the band structure of FeNbO4 is indeed revealed in the TL studies described below. | ||
| Fig. 6 Photoluminescence spectra of (a) FN-8, (b) FN-6, (c) Pt/FN-6, and (d) Pt/FN-8 photocatalysts. Excitation wavelength = 420 nm. | ||
3.6 Mott–Schottky analysis
Fig. 7 shows the plot of 1/C2 against applied dc potential. The positive slope of the M–S plot is in accordance with the n-type semiconducting nature of FeNbO4. The intercept of the linear region of this plot gives the flat band potential value, using the expression Vint = Vfb + KbTe−1, where Vint is the potential at the intercept at X-axis, Vfb is flat band potential, Kb is Boltzmann constant, T is temperature, and e is the elementary electronic charge with respect to the reference electrode.22 For an n-type material, the flat band potential can be correlated with its conduction band potential. Based on the Vint values shown in Fig. 7, the flat band potential value is estimated to be −0.45 V (considering KbTe−1 = 0.03) with respect to the normal hydrogen electrode (NHE) potential. As the electrochemical measurements were conducted at pH ∼ 13.8, extrapolation of these potentials to the actual pH of the reaction medium (∼6.5) leads to a flat band potential of −0.02 V.23 Notably, the optical bandgap of ∼2.0 eV at the absorption edge wavelength (Fig. 4) and the corresponding positive value of the conduction band potential of o-FeNbO4 (Table 3) are not in agreement with the results of the photoelectrochemical measurements. Therefore, a negative conduction band potential, as observed in M–S plot (Fig. 7), may in fact correspond to a bandgap value of >3 eV (Table 3). This is in conformity with the UV-Vis results presented in Fig. 5. | ||
| Fig. 7 Mott–Schottky plot for FN-8 photocatalyst, recorded at pH = 13.8. | ||
3.7 I–V data
Fig. 8 presents the photoelectrochemical (PEC) performance of FN-8 and a platinized FN-8 photocatalyst, measured using the aforementioned three-electrode setup. The onset potential of the photocurrent in both cases was observed around 0.1 V vs. NHE, which is higher than the flat band potential value of −0.45 V, implying a strong driving force requirement for the photogenerated charge carriers to separate and contribute towards the photocurrent. This is attributed to a high concentration of the surface states or lattice disorders in the crystal structure of FeNbO4, as indicated by the XRD, HRTEM and photo/thermo-luminescence results. The difference in the current observed under dark and light conditions contributes to the photo-induced current in FeNbO4. The observed hump at ∼0.32 V can possibly be ascribed to a change in the Fe oxidation state, as the FeNbO4 structure contains mixed Fe2+ and Fe3+ valencies.13,14 Another important feature of the I–V plots shown in Fig. 8 is the rather small photocurrent density observed for bare FeNbO4 (∼2 to 3 μA cm−2). As a benchmark, the corresponding value for a Degussa P-25 TiO2 photocatalyst with a band gap of 3.2 eV was measured to be ∼20–30 μA cm−2 under identical conditions. Thus, a low photocurrent density of FeNbO4 suggests the availability of a smaller cross section of photogenerated charge carriers under 1 sun illumination, which contains less than 7% of the UV spectrum (with wavelengths below 350 nm). Furthermore, a marginally higher photocurrent density was observed for Pt/FeNbO4 (ca. 7 μA cm−2), indicative of a more efficient charge carrier separation due to support → metal electron transfer processes. | ||
| Fig. 8 I–V plots for FN-8 and Pt/FN-8 photocatalysts. | ||
3.8 Inter-bandgap charge trapping states
Fig. 9 exhibits comparative thermoluminescence (thermally stimulated luminescence, TL) emission from FN-6 (curve a) and FN-8 (curve b) photocatalysts recorded after a lapse of ∼10 min subsequent to 1 h UV-irradiation. Curve (c) shows the TL glow emission from Pt/FN-8 under the identical experimental conditions. The TL glow curves in Fig. 9 display three main emission bands, with considerable overlap at temperature maxima (Tmax) of ca. 125, 210, and 315 °C. The energy of activation associated with these TL glow peaks (Ea ∼ 25kTmax, where k is Boltzmann's constant) is estimated to be 0.86, 1.05, and 1.26 eV. The wide (∼35 to 60 °C) TL glow peaks in Fig. 9 and the associated Ea values mentioned above reveal the existence of very closely spaced charge trapping states in between the valence band and the conduction band of FeNbO4. Another noticeable feature of these TL results is the preparation method-dependent change in the relative intensity of different glow peaks. The light emission during the TL process originates from the thermal release of e−/h+ charge pairs and their subsequent recombination at lattice/surface imperfections of inorganic semiconductors. Accordingly, the lower intensity of the TL glow emission from FN-6 (curve a) as compared to FN-8 (curve b) can be attributed to the inhibited transfer and radiative recombination of the charge pair, because of the inherent surface defects in smaller crystallites in FN-6 (Table 1). On the other hand, a significant increase in the TL intensity from Pt/FN-8 (curve c) provides clear evidence for the improved charge transfer/separation at Pt/support interfaces, in agreement with the aforementioned PEC study. Furthermore, the TL glow peaks in the temperature region of 75–350 °C in Fig. 9, associated with the energy between 1 to 1.5 eV, reveal a wide spread of the sub-bandgap charge trapping states and their close vicinity to the valence or conduction band of FeNbO4. | ||
| Fig. 9 Thermoluminescence glow curves of (a) FN-6, (b) FN-8, and (c) Pt/FN-8 photocatalysts, recorded at a heating rate of 1 °C s−1 following 1 h UV irradiation. | ||
It should be noted that no TL glow emission was observed from the samples exposed to visible light prior to thermal activation. This crucial observation highlights the vital requirement of UV light (>3 eV energy) for the bandgap excitation of FeNbO4, resulting in the generation of e−/h+ charge pairs. This observation confirms the ∼3.4 eV electronic band gap of FeNbO4.
3.9 Electronic structure
Fig. 10 presents the results of first principles electronic structure calculations on o-FeNbO4. The spin polarized band structure shows up-spin and down-spin bands separately. Noticeably, the conduction band separates the valence band by a direct band gap at gamma point (Γ) of ∼3.2 eV. The width of the valence band is about 6 eV. The dispersions of up-spin electrons are different from down-spin electrons, implying that the material is magnetic. Further, the ferromagnetic ground state is more stable than non magnetic and antiferromagnetic states by 184.99 meV and 178.38 meV respectively. The Fe atom in the unit cell has a magnetic moment of 4.76 μB. The O–Fe–O bond angle in this material is ∼94.7 degrees, in agreement with the Kanamori–Goodenough–Anderson rules, according to which a 90 degrees super-exchange interaction (magnetic ion-ligand-magnetic ion) is identified as ferromagnetic.24 Furthermore, the density of states at Fermi level is dominated by partially filled Fe-d orbital and O-p orbital, as expected from O–Fe–O super-exchange interaction. An important point to be noted in Fig. 10 is a significant contribution from Nb-d orbital at the Fermi level. Moreover, the valence band comprises of O (2p)–Nb (4s-4p)–Nb (4d-5s) orbitals and the Fe (3d) levels do not hybridize with Nb (5s) level and lie near the Fermi level.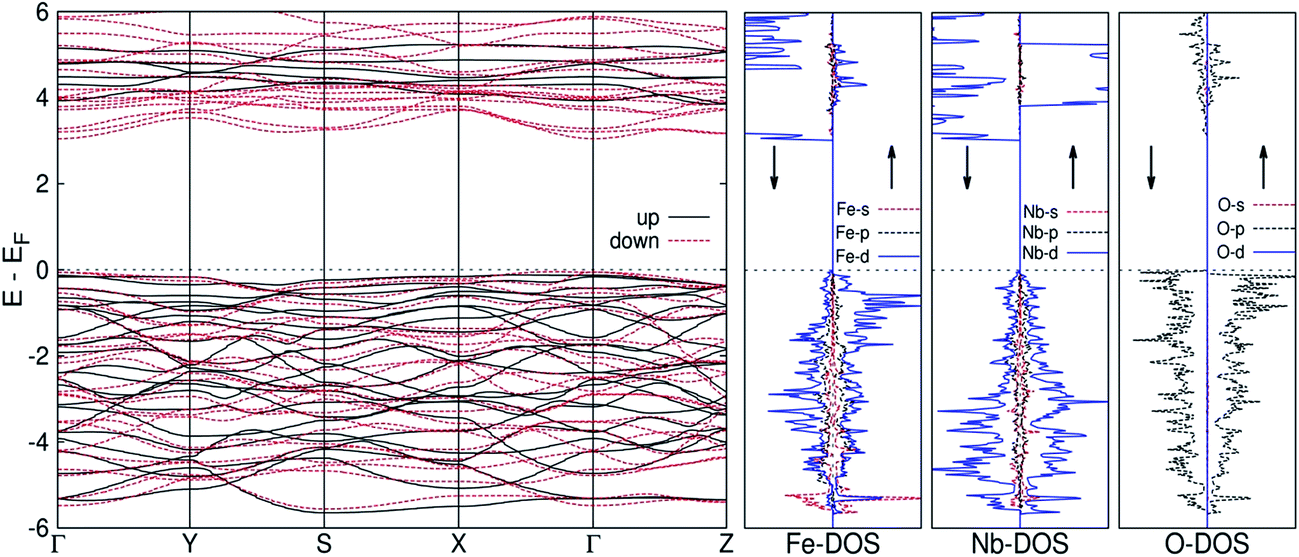 | ||
| Fig. 10 Electronic structure of o-FeNbO4, showing partial and total density of states. The zero of the energy scale refers to the Fermi energy. | ||
3.10 Photocatalytic activity
No measurable reaction occurred when an aqueous suspension of FeNbO4 photocatalyst was exposed to a 300 W xenon lamp, without or with dispersed metal co-catalysts. Similarly, no H2/O2 was evolved with the addition of sacrificial electron donors or electron acceptors and a 300 W Xe lamp for irradiation. Again, no water dissociation was observed during the irradiation of co-catalyst-free FeNbO4 samples with a high intensity solar simulator (500 W Xe-arc lamp), with and without CH3OH or AgNO3 as sacrificial reagents. However, a significant amount of O2 was evolved from the suspension of Pt/FeNbO4 and NiO/FeNbO4 photocatalysts in 10 mmol AgNO3 when illuminated with a high intensity solar simulator (500 W Xe-arc lamp). The O2 yield increased as a function of time and was observed during 2–3 consecutive reaction cycles, though with a non-reproducibility in O2 yield to the extent of ca. ±10%. Furthermore, the rate of oxygen evolution was higher for photocatalysts calcined at 600 °C and for NiO-loaded samples. The comparative O2 evolution rates obtained for different photocatalysts during visible light (>395 nm) irradiation, estimated at the end of a typical ∼5 h test run, are plotted in Fig. 11. Moreover, considerably higher O2 evolution rates were observed without a cut off filter, i.e., on illumination with the full wavelength range of the solar illuminator. Additionally, under identical reaction conditions, no reaction occurred while using CH3OH, with water![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :methanol volume ratios up to 2:1.
:methanol volume ratios up to 2:1.
 | ||
| Fig. 11 O2 evolution rate from photooxidation of water over as-synthesized (column 1) and cocatalyst-loaded FeNbO4 photocatalysts under visible light (>395 nm) illumination from solar simulator and in presence of AgNO3 as electron scavenger. | ||
4. Discussion
The main observations of this study, viz. the requirement of a strong driving force for the photogeneration of the charge carriers along with a small photocurrent density of FeNbO4 (∼2 to 3 μA cm−2, Fig. 8), requirement of a high radiation flux for O2 evolution during activity measurements, and the evolution of oxygen and no hydrogen during visible light excitation with a 500 W Xe lamp (Fig. 11), can be rationalized by considering the energy gap dependent band potentials of FeNbO4 (Table 3). The results in Table 3 show that the conduction band potential of FeNbO4 is a positive value with respect to the reduction potential of H+ to H2 for the band gap value of less than ∼3.4 eV. Accordingly, a −ve value of flat band potential obtained in our electrochemical measurements supports a bandgap energy of >3 eV for FeNbO4. It is therefore apparent that the energy gap at the wavelength of absorption edge (∼2 eV) may not represent the true valence band-to-conduction band energy gap. We suggest that the UV-visible spectra and multiple energy gaps shown in the Tauc equation plot in Fig. 5 may actually represent two distinct phenomena, one corresponding to the band-to-band (electronic) excitation (>3 eV), and another to the sub-band gap transitions giving rise to optical absorption at lower energies (<3 eV). In tune with this interpretation, Enache et al.25 attributed the strong visible light absorption of InVO4 to the presence of high density of ionized deep donor states in the space charge region of a material. Our results further support the study by Koenitzer et al.,26 where FeNbO4 was reported to have multiple energy gaps between 2.0 to 4.3 eV, originating from distinct [FeO6] and [NbO6] photoactive centres. Moreover, Blasse and 'tLam17 demonstrated that the wavelength tail in the absorption spectrum of GaNbO4 at >250 nm can be attributed to the niobate groups belonging to various local defect regions in lattice; as the local defect region becomes more extended, the corresponding wavelengths become longer. Accordingly, in contrast to the reported value of ∼2–2.2 eV,7,8 the true valence band to conduction band energy gap of o-FeNbO4 is to be taken at ∼3.4 eV. These inferences find a strong support in the electronic structure of o-FeNbO4 (Fig. 10), revealing a band-to-band energy gap of ca. 3.2 eV.The origin of the higher wave length (>400 nm) optical absorption of FeNbO4 (Fig. 5) may be traced to certain transitions involving closely spaced lattice defect-induced sub-band gap energy states. The distortion in a metal oxide crystal lattice, arising due to certain extraneous aliovalent metal impurities in addition to inherent oxygen non-stoichiometry, are known to serve as localized charge donor and charge acceptor states with an energy in between the valence band and conduction band of a metal oxide semiconductor.27 The crystal structure of FeNbO4 (Fig. 3) and the lattice parameters listed in Table 2 indeed reveal the extent of preparation-dependent disorder in the samples. As shown in Table 2, the typical (Fe/Nb)–O bond lengths (Å) are: (Fe/Nb)–O = 1.882(3) × 2; (Fe/Nb)–O = 2.098(4) × 2 and (Fe/Nb)–O = 2.045(4) × 2. Notably, the bond lengths and unit cell parameters observed were lower than those reported in the literature for o-FeNbO4 samples prepared by conventional solid state reactions and quenching methods.13 Furthermore, the bond length distortion of 20.9 × 10−4 was marginally larger than the reported value of 16.3 × 10−4 for a high temperature phase.13 This contraction of the unit cell and the comparatively higher level of distortion in the FeNbO4 lattice can be ascribed to an inherent non-equilibrium state of the structure.
Furthermore, Raman spectroscopy provides clear information about the local structure and associated distortion around different cations. The Raman frequencies associated with vibrational modes of NbOn polyhedra (n = 4 or 6) are known to depend on the bond order and the extent of distortion in the niobium oxide structure.28 The stretching modes in various tetrahedral NbO4 and octahedral NbO6 are observed at around 787 and 586 cm−1, respectively.7,29 The equilibrium Nb–O bond lengths in NbO4 tetrahedra and NbO6 octahedra are 1.823 and 1.977 Å, respectively. Per an empirical relation between the stretching frequency and Nb–O bond length for different coordination polyhedral (ν(cm−1) = 25922e(−1.9168×R), where ν is frequency of the stretching mode and R is Nb–O bond length in Å),7 the bond lengths corresponding to the frequencies at 816 and 604 cm−1 of FNb-8 are 1.80 and 1.96 Å, respectively. A significant difference observed in the Fe/Nb–O bond lengths, 1.88 to 2.10 Å from the XRD data and the corresponding values of 1.80 to 1.96 Å from the Raman study indicate a larger dispersion in the bond lengths. This can be attributed to the intrinsic distortion in the octahedral units. Furthermore, the Raman modes due to the stretching frequencies from Fe–O bonds in FeO6 octahedra are expected at around 407 and 605 cm−1.30 Since the ionic radii of Fe3+and Nb5+ are similar, a significant difference between the Fe–O and Nb–O bond lengths is therefore unexpected. Thus, the Raman spectra in Fig. 4 indicate a significant distortion in the octahedral units of o-FeNbO4. Furthermore, the broadening of the Raman lines and a shift in their peak position, as observed in case of Raman spectrum of FN-6 compared to FN-8 (Fig. 4b–d), have been attributed earlier to various factors, such as: disruption of long range ordering in lattice structure, composition fluctuations, particle size-dependent phonon confinement, non-stoichiometry, or internal stress/surface tension effects at metal/substrate interfaces.6,31
Highlighting the above-described findings of our study, Fig. 12 presents a schematic illustration of the electronic versus optical band characteristics of FeNbO4 along with the energy transitions responsible for PL (Fig. 6) and TL (Fig. 9) emissions from photo-excited photocatalysts. The bandgap energy and the lattice distortion-induced inter-bandgap energy states responsible for visible light absorbance, as depicted in Fig. 12, find strong support from our XRD (Fig. 2 and 3), laser-Raman (Fig. 4) and the electronic structure (Fig. 10) investigations described above. Moreover, a wide variation in the particle size ranges in our samples (Table 1) may also play a role in the multiple absorption states shown in Fig. 5 (inset).
 | ||
| Fig. 12 A schematic diagram showing possible energy transitions in FeNbO4 (electronic bandgap ∼3.4 eV, optical bandgap ∼2, 2.4 eV), photo-absorption leading to e−/h+ pair generation (step i), or entrapment of charge carriers at defect-induced sub-bandgap trapping states (step ii). Step (iii) shows thermal- or photo-assisted de-trapping of charge carriers, and step (iv) their subsequent recombination leading to photo- or thermo-luminescence emission. Step (v) represents a band-to-band excitonic recombination. | ||
The cocatalyst-dependent O2 evolution rates from visible-light irradiation of metal/FeNbO4 composites, presented in Fig. 11, bear a relationship with particle morphology and microstructure (Fig. 1 and 4). Thus, the superior activity of FN-6, comprising smaller size particles compared to FN-8 (Table 1), is in conformity with our earlier studies on particle size effects in photocatalytic activity,32,33 even though no quantitative relationship could be established. As is well reported, the small size particles provide not only a large surface to volume ratio but also certain coordinative unsaturated surface sites, which in turn translate to an enhanced number of active reaction sites and influence the dynamics of charge-carrier transfer.34 Moreover, the selective exposure of certain lattice planes, as shown in Fig. 1e and f, may play an important role in observed catalytic activity. The importance of facet engineering in controlling of photocatalytic activity, where specific surface sites exhibit distinct adsorption characteristics, is now being realized increasingly.35,36 Further, a comparatively higher photocurrent density (∼7 μA cm−2), low intensity PL emission (Fig. 6), and wide Laser Raman spectral bands (Fig. 4) for Pt/FeNbO4 are attributed to well-reported role of dispersed metal co-catalysts in e−/h+ charge separation, leading thereby to an enhanced catalytic activity (Fig. 11).6,18 We have discussed earlier in detail how the finely dispersed metal nanoparticles and the architecture of metal/oxide heterojunctions (TEM evidence) may govern the photocatalytic activity of a metal/semiconductor nanocomposite.33,37 In conclusion, the present study reaffirms that the photocatalytic activity of a wide-gap semiconductor may depend on a combination of several factors, viz. electronic structure, grain morphology, exposure of a particular lattice plane, surface and metal/semiconductor interfacial properties, and most importantly, the presence of certain size- and structure-dependent microstructural lattice disorders.
5. Summary
The following points represent the main findings of our study:1. Orthorhombic phase FeNbO4, prepared hydrothermally, shows a higher level of lattice distortion compared to that prepared by solid-state methods. This is ascribed to the restricted migration of cations at low calcination temperatures (≤800 °C), and the excessive contribution of surface energy associated with nanosized particles (Table 1).
2. The first principles electronic structure of o-FeNbO4, calculated from a modelled structure of FeNbO4, is reported here for the first time. The o-FeNbO4 exhibits a stable ferromagnetic state, where the conduction band separates the valence band by a direct band gap of ∼3.2 eV at gamma point (Γ).
3. The o-FeNbO4 displayed a multistep optical absorbance attributed to at least two distinct absorbance regimes: one in the UV region due to band gap energy (∼3.4 eV) excitation and the other in the visible region due to the transitions involving inter-bandgap donor or acceptor energy states. The fact that TL emission was observed only for UV excitation and not after exposure to visible light supported the ∼3.4 eV electronic bandgap of FeNbO4.
4. The PEC measurements further confirm the wide bandgap for FeNbO4. The PL, TL, and laser-Raman studies revealed the presence of certain localized and close-spaced charge trapping states, with energies in between the valence band and conduction band potentials of FeNbO4.
5. In tune with a +ve value of the flat band potential with respect to the redox potential of H+/H2 (0 V vs. NHE) for an optical band gap of 2–2.4 eV (Fig. 10, Table 3), no H2 evolution was observed during visible light irradiation of FeNbO4 even in the presence of an electron donor. A significant activity for visible light photooxidation of water was however observed using AgNO3 as an electron scavenger. The rate of O2 evolution increased considerably upon dispersion of a metal co-catalyst, more so for NiO. The photoactivity of FeNbO4 was controlled by multiple factors, such as: band characteristics, grain morphology and microstructure.
6. Contrary to prevalent assumptions, the wavelength at the onset of the optical absorption edge may not represent the true valence band-to-conduction band energy gap (electronic band gap) of metal oxide semiconductors.
Acknowledgements
The financial support for this research is provided by the Science and Engineering Research Council (SERC, Chemical Engineering) of the Department of Science and Technology, New Delhi. Some of the sample characterizations were performed at the Center for Material Characterization (CMC) at NCL. The TL data on our samples were provided by Dr M. S. Kulkarni of Bhabha Atomic Research Center, Mumbai. Ms Suman Devi and Ms Aparna Deshpande are thanked for their help.References
- Y. Miseki and A. Kudo, ChemSusChem, 2011, 4, 245 CAS.
- O. C. Compton and F. E. Osterloh, J. Phys. Chem. C, 2009, 113, 479 CAS.
- J. Ye, Z. Zou, H. Arakawa, M. Oshikiri, M. Shimoda, A. Matsushita and T. Shishido, J. Photochem. Photobiol., A, 2002, 148, 79 CrossRef CAS.
- L. Wang, W. Wang, M. Shang, S. Sun, W. Yin, J. Ren and J. Zhou, J. Mater. Chem., 2010, 20, 8405 RSC.
- Z. Zou and H. Arakawa, J. Photochem. Photobiol., A, 2003, 158, 145 CrossRef CAS.
- S. Devi, S. Kelkar, V. Kashid, H. G. Salunke and N. M. Gupta, RSC Adv., 2013, 3, 16817 RSC.
- I.-S. Cho, S. Lee, J. H. Noh, G. K. Choi, H. S. Jung, D. W. Kim and K. S. Hong, J. Phys. Chem. C, 2008, 112, 18393 CAS.
- H. Leiva, K. Sieber, B. Khazai, K. Dwight and A. Wold, J. Solid State Chem., 1982, 44, 113 CrossRef CAS.
- P. Dhak, D. Dhak, M. Das, T. Subhashchandrabose and P. Pramanic, J. Nanopart. Res., 2011, 13, 4153 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15 CrossRef CAS; G. Kresse and J. Furthmüller, Phys. Rev. B: Condens. Matter Mater. Phys., 1996, 54, 11169 CrossRef.
- W. Q. Fang, X.-Q. Gong and H. G. Yang, J. Phys. Chem. Lett., 2011, 2, 725 CrossRef CAS.
- O. Raymond, R. Font, N. Suarez, J. Portelles and J. M. Siqueiros, Ferroelectrics, 2003, 294, 141 CAS.
- W. T. A. Harrison and A. K. Cheetham, Mater. Res. Bull., 1989, 24, 523 CrossRef CAS.
- E. Schmidbauer and J. Schneider, J. Solid State Chem., 1997, 134, 253 CrossRef CAS.
- A. C. Larson and R. B. Von Dreele, GSAS: General Structure Analysis System, Los Alamos National Laboratory, report LA-UR 86-748, 2000 Search PubMed.
- G. Blasse, J. Solid State Chem., 1973, 7, 169 CrossRef CAS.
- G. Blasse and R. U. E. 'tLam, J. Solid State Chem., 1978, 25, 77 CrossRef CAS.
- U. Pal and P. Santiago, J. Phys. Chem. B, 2005, 109, 15317 CrossRef CAS PubMed.
- J. Yang, D. Wang, H. Han and C. Li, Acc. Chem. Res., 2013, 46, 1900 CrossRef CAS PubMed.
- C. H. Liang, G. W. Meng, Y. Lei, F. Phillipp and L. D. Zhang, Adv. Mater., 2001, 13, 1330 CrossRef CAS.
- S. J. Chen, Y. C. Liu, C. L. Sho, R. Mu, Y. M. Lu, J. Y. Zhang, D. Z. Shen and X. W. Fan, Adv. Mater., 2005, 17, 586 CrossRef CAS.
- K. Gelderman, L. Lee and S. W. Donne, J. Chem. Educ., 2007, 84, 685 CrossRef CAS.
- T. Bak, J. Nowotny, M. Rekas and C. C. Sorrell, Int. J. Hydrogen Energy, 2002, 27, 991 CrossRef CAS.
- J. Kanamori, J. Phys. Chem. Solids, 1959, 10, 87 CrossRef CAS , and back references.
- C. S. Enache, D. Lloyd, M. R. Damen, J. Schoonman and R. Van de krol, J. Phys. Chem. C, 2009, 113, 19351 CAS.
- J. Koenitzer, B. Khazai, J. Hormadaly, R. Kershaw, K. Dwight and A. Wold, J. Solid State Chem., 1980, 35, 128 CrossRef CAS.
- T. Bak, J. Nowotny, M. Rekas and C. C. Sorrell, J. Phys. Chem. Solids, 2003, 64, 1057 CrossRef CAS.
- J.-M. Jehng and I. E. Wachs, Chem. Mater., 1991, 3, 100 CrossRef CAS.
- F. D. Hardcastle and I. E. Wachs, Solid State Ionics, 1991, 45, 201 CrossRef CAS.
- D. L. A. De Faria, S. V. Silva and M. T. De Oliveira, J. Raman Spectrosc., 1997, 28, 873 CrossRef CAS.
- V. Y. Davydov, I. N. Goncharuk, A. N. Smirnov, A. E. Nicolaev, W. V. Lundin, A. S. Usikov, A. A. Klochikhin, J. Aderhold, J. Graul, O. Semchinova and H. Harima, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 125203 CrossRef.
- P. Dhanasekaran and N. M. Gupta, Mater. Res. Bull., 2012, 47, 1217 CrossRef PubMed.
- S. V. Awate, S. S. Deshpande, K. Rakesh, P. Dhanasekaran and N. M. Gupta, Phys. Chem. Chem. Phys., 2011, 13, 11329 RSC.
- D. Beydoun, R. Amal, G. Low and S. McEvoy, J. Nanopart. Res., 1999, 1, 439 CrossRef CAS.
- N. Li, M. Liu, Z. Zhou, J. Zhou, Y. Sun and L. Guo, Nanoscale, 2014 10.1039/c4nr02068e.
- W. Jiao, L. Wang, G. Liu, G. Q. Lu and H. M. Cheng, ACS Catal., 2012, 2, 1854 CrossRef CAS.
- P. Dhanasekaran and N. M. Gupta, Int. J. Hydrogen Energy, 2012, 37, 4897 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |