
An investigation of physico-chemical properties of a new polyimide–silica composites
T. Akhtera,
O. Ok Parka,
H. M. Siddiqi*b,
S. Saeedcd and
Khaled Mohammad Saoudd
aDepartment of Chemical and Biomolecular Engineering (BK21+ Graduate Program), KAIST, 291 Daehak-ro, Yuseong-gu, Daejeon 305-701, Republic of Korea
bDepartment of Chemistry, Quaid-i-Azam University, Islamabad 45320, Pakistan
cDepartment of Metallurgy and Materials Engineering, Pakistan Institute of Engineering and Applied Sciences (PIEAS), Islamabad 45650, Pakistan
dDepartment of Liberal Arts and Sciences, Virginia Commonwealth University in Qatar, PO Box 8095, Doha, Qatar
First published on 1st September 2014
Abstract
In the present study, a novel diamine 1,4-bis[4-(hydrazonomethyl)phenoxy]butane (4-BHPB) has been successfully synthesized by a facile method. 4-BHPB was reacted with pyromellitic dianhydride (PMDA) to derive a novel polyimide (PI). Highly compatibilized PI–SiO2 nanocomposites were tailored using synthesized PI matrix and modified silica nanoparticles. The compatibility between organic–inorganic (O–I) components was remarkably improved by charge transfer complex (CTC) formed between PI chains and modified silica nanoparticles. PI chains have electron donor and acceptor groups (diamine and dianhydride portion, respectively). These groups were generated in silica nanoparticles by the organic modification through 2,6-bis(3-(triethoxysilyl)propyl)pyrrolo[3,4-f]isoindole-1,3,5,7(2H,6H)-tetraone (M-SiO2), which in turn, was prepared reacting PMDA with 3-aminopropyltriethoxysilane. Sol–gel method was used for in situ synthesis of silica nanoparticles from a mixture of M-SiO2 and TEOS. The enhanced compatibility between O–I phases through CTC formation furnished PI–SiO2 nanocomposites (designated as OI-M) with improved thermal stability, hydrophobicity, and surface smoothness. For the comparison of properties, another series of PI–SiO2 composites (OI-UM) were prepared dispersing unmodified silica micro-particles into PI matrix. OI-UM hybrid system does not contain CTC between PI matrix and silica particles. The structure of the monomer, PI, and organically modified silica network was analyzed by FTIR, and NMR spectroscopy. Thermogravimetric analysis, FE-SEM, contact angle measurement, and AFM were used to study thermal and morphological properties of the synthesized PI–SiO2 hybrids.
Introduction
Polyimides (PIs) have attracted substantial importance as coating materials in microelectronics, aircraft, and electrical industries because they exhibit high mechanical and thermal stability, good optical properties, excellent resistance against organic solvents.1–4 However, certain applications like waveguide materials, spacecraft, electronics, and aircraft industries demand more improvements in hydrophobicity, thermal properties, and surface smoothness.5 The properties and performance of PIs may be improved further by the addition of nano-scale silica into PI matrix giving organic–inorganic (O–I) hybrid materials.6–9 O–I hybrids merge together the synergistic properties of PI matrix (ductility, and low temperature processing) with low coefficient of thermal expansion and high thermal stability of silica nanoparticles.10–12 A number of techniques have been attempted for the preparation of O–I hybrid.1,2,13 Among these techniques, sol–gel reaction is the most widely explored method for the preparation of O–I hybrids because of its versatility and provision of better control over surface morphology.12,14–17 However, inorganic silicates and organic polymers behave differently during the condensation process of sol–gel reaction, as a result O–I phases are segregated from each other.4,18–20 Properties of O–I composites are believed to be the result of interfacial interaction between matrix and filler phases.21–24 A number of research efforts are reported to avoid or at least minimize the phase separation between O–I components by adding different coupling agents.12,25–30 Various researchers studied the surface modification of silica particles to make them more compatible with organic matrix. Their studies demonstrated the improvement in the properties of O–I hybrid materials.9,15 For instance, Musto et al.10 explored the effect of controlled particle size and improved morphology on the properties of PI–SiO2 composites prepared via sol–gel method by introducing γ-glycidoxypropyltrimethoxysilane as interphase coupling agent. Similarly, enhanced thermal and mechanical properties of O–I hybrids were achieved by Khalil et al.31 using aminophenyltrimethoxysilane as a coupling agent owning to controlled particles size and uniform dispersion of nano-scale silica phase into PI matrix.Many researchers studied the tendency of PI chains to form charge transfer complex (CTC) with other polymer chains through their electron donor (diamine portion) and electron acceptor groups (dianhydride).32–34
The present study reports the improvement of compatibility between polyimide matrix and silica nanoparticles through CTC. For this purpose a new diamine 1,4-bis[4-(hydrazonomethyl) phenoxy]butane (4-BHPB) was synthesized. 4-BHPB was subsequently reacted with pyromellitic dianhydride (PMDA) to prepare a novel PI. The PI chain inherently possesses electron-donor groups (diamine portion) and electron-acceptor groups (dianhydride portion). Existence of electron-donor and -acceptor groups in the reinforcement and thereby formation of CTC with PI chains was confirmed by the organic modification of SiO2 nanoparticles. Silica nanoparticles were synthesized using a mixture of TEOS and 2,6-bis(3-(triethoxysilyl)propyl) pyrrolo[3,4-f]isoindole-1,3,5,7(2H,6H)-tetraone (M-SiO2). M-SiO2 was prepared by reacting APrTEOS with PMDA. Subsequently, synthesized PI–SiO2 nanocomposites (designated as OI-M) exhibited significantly improved thermal, morphological, and hydrophobic properties owing to enhanced phase connectivity between PI matrix and silica nanoparticles, which is a direct consequence of CTC formed between O–I components. Moreover, PI matrix and silica nanoparticles both have alkyl groups, which may enhance the interfacial interaction between two phases through nonpolar interactions. For the evaluation of property enhancement and comparative study, a series of PI–SiO2 composites (OI-UM) was also prepared where the unmodified silica network was dispersed into PI matrix and resulting composites lack in CTC between O–I components. The percentage of silica phase in both hybrid system (OI-M and OI-UM) was varied from 0–30 wt% and their properties were studied using appropriate analytical techniques.
Experimental
Materials
4-Hydroxybenzaldehyde (Merck, Germany), 1,4-dibromobutane (Merck, Germany), potassium carbonate (Merck, Germany), ethanol (Merck, Germany), hydrazine monohydrate (64–65%, Sigma-Aldrich, USA), dimethylformamide (DMF, Merck, Germany), 4,4′-oxydianiline (ODA, 97%, Sigma-Aldrich, USA), pyromellitic dianhydride (PMDA, 99.2%, Sigma-Aldrich, USA), dimethylacetamide (DMAc, 99.8%, water content <0.005%, Sigma-Aldrich, USA), tetraethoxysilane (TEOS, 97.5%, Acros Organics, Fair Lawn, N J), and 3-aminopropyltriehoxysilane (APrTEOS, 95%, ABCR GmbH & Co, Germany).Synthesis
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1), the reaction mixture was cooled to room temperature. Allowing the reaction mixture to stand for 1 h resulted in the precipitation of crude product, which was retrieved by filtration. The dried product was re-dissolved in THF. The final product was precipitated by cooling the solution to 0 °C and was separated by filtration.
:1), the reaction mixture was cooled to room temperature. Allowing the reaction mixture to stand for 1 h resulted in the precipitation of crude product, which was retrieved by filtration. The dried product was re-dissolved in THF. The final product was precipitated by cooling the solution to 0 °C and was separated by filtration.
 | ||
| Fig. 1 Synthesis of 1,4-bis[4-(hydrazonomethyl)phenoxy]butane (4-BHPB). | ||
Yield: 78%. m.p 262–263 °C; FTIR: ν− (cm−1) 3387, 3347 (NH2), 2942, 2863 (–CH2), 1623 (–CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N). 1H NMR (300 MHz, DMSO-d6): δ ppm 7.65 (1H, s, methine proton), 6.52 (4H, s, NH2), 4.02 (4H, s, 5, 5′), 1.85 (4H, s, 6, 6′), 6.91–6.88 (4H, d, J = 8.7 Hz, 2, 2′), 7.41–7.38 (4H, d, J = 8.7 Hz, 3, 3′); 13C NMR (75 MHz, DMSO-d6): δ ppm 129.45 (1), 114.96 (2, 2′), 126.96 (3, 3′), 158.70 (4), 67.56 (5, 5′), 25.87 (6, 6′) 139.17 (CN). Anal. calcd for C18H22N4O2: C, 66.24; H, 6.79; N, 17.17. Found: C, 66.04; H, 6.45; N, 16.99.
N). 1H NMR (300 MHz, DMSO-d6): δ ppm 7.65 (1H, s, methine proton), 6.52 (4H, s, NH2), 4.02 (4H, s, 5, 5′), 1.85 (4H, s, 6, 6′), 6.91–6.88 (4H, d, J = 8.7 Hz, 2, 2′), 7.41–7.38 (4H, d, J = 8.7 Hz, 3, 3′); 13C NMR (75 MHz, DMSO-d6): δ ppm 129.45 (1), 114.96 (2, 2′), 126.96 (3, 3′), 158.70 (4), 67.56 (5, 5′), 25.87 (6, 6′) 139.17 (CN). Anal. calcd for C18H22N4O2: C, 66.24; H, 6.79; N, 17.17. Found: C, 66.04; H, 6.45; N, 16.99.
 | ||
| Fig. 2 Synthesis of PI. | ||
:1) in DMAc for 4 h. Subsequently, an appropriate amount of TEOS (40% solution in DMAc) was added and the oligomeric solution was stirred further for 1 h.
 | ||
| Fig. 3 Synthesis of M-SiO2. | ||
Synthesis of polyimide-unmodified SiO2 composite (OI-UM). In OI-UM hybrid system, unmodified silica micro-particles were dispersed in situ in PI matrix. An appropriate amount of TEOS (40 wt% solution in DMAc) was added to the PAA solution as given in Table 1. The reaction mixture was stirred for 1 h followed by the addition of acidified water. The temperature of the reaction mixture was raised to 60 °C followed by stirring for another 6 h. Subsequently, the reaction mixture was transferred to Teflon petri dishes and heated at 70 °C for 12 h to cast OI-UM hybrid films. The resultant flexible films were thermally imidized at 100 °C, 200 °C, and 300 °C each for 1 h.
| PI–SiO2 composites | Codes | M-SiO2:TEOS (%) |
Film composition | Film | |
|---|---|---|---|---|---|
| PI (wt%) | SiO2 (wt%) | ||||
| a F = flexible, T = transparent, O = opaque. | |||||
| Pristine PI | 0-PI | 00:00 |
100 | 0 | F, T |
| OI-UM | |||||
| 10UM | 0:1 |
90 | 10 | F, O | |
| 20UM | 0:1 |
80 | 20 | F, O | |
| 30UM | 0:1 |
70 | 30 | F, O | |
| OI-M | |||||
| 10M | 0.05:0.95 |
90 | 10 | F, T | |
| 20M | 0.05:095 |
80 | 20 | F, T | |
| 30M | 0.05:0.95 |
70 | 30 | F, T | |
Synthesis of polyimide-modified SiO2 composite (OI-M). The OI-M hybrid system was prepared by in situ generation of modified silica nanoparticles. A mixture of M-SiO2, PAA solution, and acidified water was subjected to sol–gel reaction. Next, the solvent elution technique and thermal imidization process were exercised to cast and imidized hybrid films, respectively, as discussed earlier.
0–30 wt% SiO2 was incorporated into the PI matrix (both in OI-UM and OI-M) via sol–gel reaction. Table 1 presents the compositions and codes of hybrid films of OI-M and OI-UM composites.
Characterization
1H and 13C NMR spectroscopy. Structures and purity of the synthesized monomer were elucidated by 1H and 13C NMR spectroscopy. Bruker AM-300 spectrophotometer was used to record 1H and 13C NMR spectra at 300 MHz and 75 MHz, respectively.
Solid state 29Si NMR spectroscopy. The 29Si NMR spectra, recorded on Bruker Advance 400WB NMR Spectrometer, were used to investigate the structure of organically modified silica network. The hybrid films were ground to powder form in freeze drier. Various silicate nuclei were differentiated from each other using very facile nomenclature. The silicate species having one organic side group were denoted by letter “T”. “Qi” were used to denote the species, which do not have any organic side group, whereas “i” stands for the number of –O–Si groups directly bonded to Si atom.
Field emission scanning electron microscopy (FE-SEM). The surface morphology of both OI-M and OI-UM hybrid films was studied using field emission scanning electron microscope S-4800. Flexible composite films were dipped in liquid nitrogen, and then were cryo-fractured. Before examining, the samples were sputter-coated with platinum and vaulted on aluminium holders with the help of carbon tape. Secondary electron images were collected with a beam voltage of 10 keV using an in-lens detector.
Atomic force microscopy (AFM). Surface roughness analysis of the hybrid films was carried out with the help of multimode digital instruments DI 3100 Nanoman AFM, Veeco Metrology, LLC, USA. Scanning probe microscopy (triangular cantilever containing a pyramidal tip made of silicon nitride) software WSxM 3.0 Beta v. 8.1 was utilized to capture AFM micrographs in taping-mode at ambient conditions.
Contact angle measurements. The contact angle of pristine PI, OI-M, and OI-UM composites was analyzed on Phoenix 300 Plus, SEO Co, Ltd Contact Angle Analyzer using deionized water as providing liquid.
Thermogravimetric analysis (TGA). TGA analysis at TG 209 F3, NETZSCH Germany, was used to access the thermal stability of neat PI and its silica reinforced composites. The vacuum dried samples were analyzed at a heating rate of 10 °C min−1 from 30 to 800 °C in an inert nitrogen atmosphere.
Results and discussion
Monomer synthesis
Fig. 1 presents the synthesis of 1,4-bis[4-(hydrazonomethyl) phenoxy]butane (4-BHPB), which comprises of two steps. The first step includes synthesis of 4,4′-(ethane-1,2-diylbis(oxy))dibenzaldehyde by the nucleophilic substitution reaction of 4-hydroxybenzaldehyde with dibromoethane. Whereas, in the second step condensation reaction between 4,4′-(ethane-1,2-diylbis(oxy))dibenzaldehyde and hydrazine monohydrate resulted in the preparation of 4-BHPB. The purity and structure of the synthesized product were comprehensively confirmed by FTIR, elemental analysis, 1H and 13C NMR spectroscopic techniques.In the FTIR spectra, the diagnostic absorption band, attributed to –CN, was observed at 1623 cm−1. The absorption bands observed at 3387 cm−1 and 3347 cm−1 were assigned to asymmetric and symmetric stretching vibrations of the amino group, respectively. Similarly, the para-substitution pattern of the benzene ring in the product was confirmed by an absorption band at 820 cm−1.
The structure of 4-BHPB was further evaluated by the 1H NMR spectroscopy where a singlet of two protons at 7.65 ppm, assigned to the protons of azomethine groups, confirmed the successful formation of the product (Fig. 4). Four protons of amino group resonated at 6.52 ppm as singlet in the spectrum. In the aliphatic region, the four protons of methylene groups (5,5′) bonded to the oxygen atom resonated at 4.02 ppm, while protons of the central methylene groups (6,6′) appeared at 1.85 ppm.
 | ||
| Fig. 4 1H HMR spectrum of 4-BHPB. | ||
Further confirmation about the structure of 4-BHPB was derived from the 13C NMR spectrum (Fig. 5) where the peak for carbon atom of azomethine group was observed at 139.17 ppm. Thus, the NMR data confirmed that the structure of 4-BHPB agreed with the proposed molecular structure.
 | ||
| Fig. 5 13C NMR spectrum of 4-BHPB. | ||
Characterization of polyimide–SiO2 composites
FTIR spectroscopy. FTIR spectroscopy was used to analyze the conversion of monomers into PAA, and PAA into PI.
FTIR spectrum of PI (Fig. 6) shows that an absorption band at around 1650 cm−1 (characteristic of amide carbonyl group in PAA) was not observed. Thus, the presence of silica network imposed no detrimental effect on PI curing, which is in good agreement with the previously published reports.21,37 Conversion of PAA into PI was further supported by the presence of characteristic absorption band for asymmetric and symmetric stretching vibration of carbonyl groups (1759 cm−1, 1720 cm−1, respectively), C–N stretching vibration (1360 cm−1), and imide ring deformation (722 cm−1).
 | ||
| Fig. 6 FTIR spectra of PI and PI–SiO2 hybrids: (a) PI, PAA (b) OI-M hybrids (c) OI-UM hybrids. | ||
In the FTIR spectra of the OI-M and OI-UM hybrid system, a broad absorption band around 1100–1020 cm−1 is present, the intensity of this broad absorption band was observed to increase as the silica contents increased from 10–30 wt%. This absorption band appeared because of the stretching vibrations of Si–O–Si linkages in the silica network.37 Moreover, it is also evident from Fig. 6 that the height of this band was smaller and located at lower wavenumbers in case of the OI-M system as compared to the OI-UM system. Thus, it may be inferred that in the OI-UM hybrid system the structure of silica network is dense and cyclic as compared to the OI-M hybrid system where more open silica networks prevailed due to preceding chemical modification.37
Solid-state 29Si NMR spectroscopy. 29Si NMR spectroscopy was used to evaluate the structure of silica network and to investigate the bonding environment of silicate nuclei. 29Si NMR spectrum was recorded using cross polarization magic angle spinning (CP-MAS) technique with a relaxation delay of 3 s. Fig. 7 presents the 29Si NMR spectrum of 10M from the OI-M hybrid system. Silica network in the OI-M hybrid system comprised of four different types of silicate nuclei i.e., Si(OSi)4 (Q4) at −110 ppm, Si(OSi)3(OH) (Q3) at −100 ppm, Si(OSi)2OH2 (Q2) at −91 ppm, and (SiO)3Si–C (T) at −65 ppm. These chemical shift values are in accordance with the published literature.38–40 The resonance peak corresponding to (SiO)3Si–C nuclei (T) exhibits the successful organic modification of silica phase in the OI-M hybrid system.9,38,39 The organic modification of silica nanoparticles resulted in uniform dispersion of particles in the PI matrix and controlling the phase segregation between O–I component imparting the advanced properties to OI-M hybrid films.
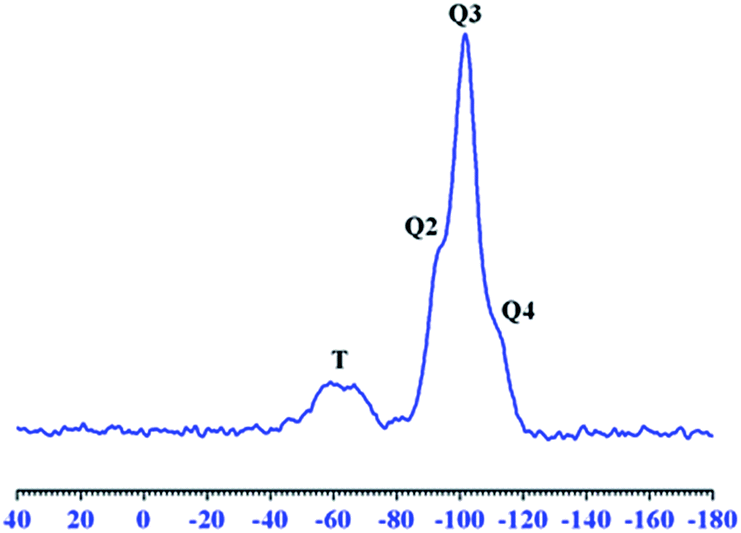 | ||
| Fig. 7 Solid-state 29Si NMR spectrum of 10M. | ||
Microstructural analysis
 | ||
| Fig. 8 FE-SEM micrographs (a) 10UM, (b) 10M, (c) 20UM, (d) 20M, (e) 20UM at 500 nm, (f) 20M at 5 μm. | ||
 | ||
| Fig. 9 Charge transfer complex and nonpolar interactions between PI chains and silica nanoparticles. | ||
 | ||
| Fig. 10 AFM micrographs (3 × 3 μm) of (a and b) 3D and 2D images of OI-UM hybrid system (c and d) 3D and 2D images of OI-M hybrid system. | ||
Contact angle measurement
Contact angle analysis furnishes useful information about the hydrophobicity or hydrophilicity of the surface of the polymer materials. Hydrophobic surfaces have contact angles in the range of about 70° to 90°, while hydrophilic surfaces have low values (0–30°).41 The values of contact angle (θ) for OI-M and OI-UM systems are given in Table 2. The variations in the contact angle as a function of silica contents are shown in Fig. 11. In the case of the OI-M, integration of silica nanoparticles into the PI matrix lead to an increase in the contact angle values from 65.5° for the pristine PI to 93.3° at 30 wt% silica loading. On the other hand, this value increased to 83.2° in the case of the OI-UM hybrid at 30 wt% silica content.| Codes | Contact angle (θ) |
|---|---|
| 0-PI | 65.5 |
| OI-UM | |
| 10UM | 74.9 |
| 20UM | 79.2 |
| 30UM | 83.2 |
| OI-M | |
| 10M | 79.6 |
| 20M | 87.3 |
| 30M | 93.3 |
 | ||
| Fig. 11 Comparison of the contact angle of the OI-M and OI-UM hybrid systems as a function of silica content. | ||
Thus, OI-M hybrid films were found more hydrophobic with an increase of 27.8° in contact angle values (0 to 30 wt%) as compared to OI-UM hybrid films, which exhibit an increment of 17.7°. Higher values of contact angle and enhanced hydrophobicity of the OI-M system as compared to OI-UM system may be attributed to nonpolar interactions of alkyl groups and CTC as a result of electron donor and electron acceptor groups present together in the O–I phases. Enhanced hydrophobicity of PI–SiO2 coatings prevent the infusion of water to the substrate (e.g. metal and/or ceramics), thereby minimizing the chances of deterioration of the coating.42
Thermal properties
The effect of improved miscibility of PI matrix with silica nanoparticles through CTC on the thermal properties of resulting hybrid films was evaluated using the thermogravimetric analysis. The hybrid films were analyzed at a heating rate of 10 °C min−1 in the inert nitrogen atmosphere. Fig. 12 shows the TGA thermograms of both hybrid systems reinforced with 10–30 wt% silica phase, whereas Table 3 gives some derived data. The thermal stability of the hybrids was assessed in terms of temperatures at 10% weight loss (T10), and residual mass at 800 °C. Table 3 shows that the value of thermal decomposition temperature T10 is 558 °C for pristine PI, while it was 20 °C higher for OI-UM and 34 °C higher for the OI-M hybrid system at 30 wt% silica content as compared to pristine PI. A comparison of the thermal stability among both the hybrid systems exhibited that modified silica nanoparticles raised the values of T10 to a greater extent as compared to the unmodified silica networks. Similarly, the residual mass at 800 °C has higher values for the OI-M hybrid system as compared to the OI-UM system. The delayed decomposition process of the PI matrix revealed that incorporation of modified silica nanoparticles resisted the diffusion of oxygen into the PI matrix and consequently reduced the degradation at elevated temperature.31 In contrast to the unmodified silica particles, the modified silica phase was well adhered and well mixed to the matrix. The existence of CTC because of the electron donor–acceptor groups and nonpolar interactions due to alkyl groups simultaneously present in the O–I phases makes organically modified silica nanoparticles effective heat and mass transport barriers; hence, conserved the matrix more efficiently.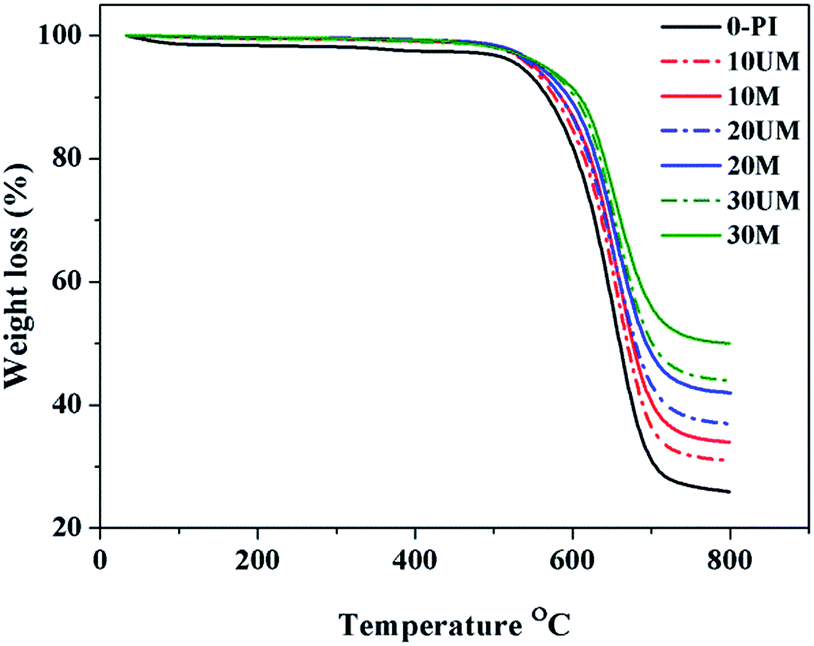 | ||
| Fig. 12 Comparison of thermal stability of OI-M and OI-UM hybrid systems. | ||
| Codes | T10 (°C) | Residual mass (wt%) |
|---|---|---|
| 0-PI | 558 | 25 |
| OI-M | ||
| 10M | 569 | 33 |
| 20M | 580 | 41 |
| 30M | 592 | 49 |
| OI-UM | ||
| 10UM | 563 | 30 |
| 20UM | 570 | 36 |
| 30UM | 578 | 43 |
Conclusions
The charge transfer complex (through novel diamine 4-BHPB in PI matrix and M-SiO2 in silica nanoparticles), as well as nonpolar interaction between the alkyl groups of O–I components significantly raised the compatibility between PI matrix and nano-scale silica phase. The presence of different types of silicate nuclei and organic modification of silica domain was investigated by solid state 29Si NMR spectroscopy. FE-SEM micrographs revealed that M-SiO2 led to controlled particle size (below 50 nm), considerably reduced agglomeration, and uniform dispersion of silica nanoparticles into the PI matrix in case of OI-M hybrids. In comparison, OI-UM hybrids displayed silica micro-beads completely detached from the PI matrix with particle size ranging between 1–2 μm. Similarly, OI-M hybrid films were found more hydrophobic as compared to OI-UM hybrid films. Thermal stability exhibited the similar trend, i.e. values of T10 and residual mass at 800 °C remained always higher for the OI-M hybrid system as compared to the OI-UM hybrid system. Thus, it can be inferred that CTC between O–I phases presented a remarkable enhancement to the eventual properties of the OI-M hybrid system by ensuring efficient dispersion of silica nanoparticles, strengthening interfacial interactions, and inhibiting phase segregation.Notes and references
- M. Miki, T. Suzuki and Y. Yamada, J. Appl. Polym. Sci., 2013, 130, 54–62 CrossRef CAS.
- T. Suzuki and Y. Yamada, J. Appl. Polym. Sci., 2013, 127, 316–322 CrossRef CAS.
- T. Akhter, S. Saeed, H. Siddiqi, O. O. Park and G. Ali, J. Polym. Res., 2013, 21, 1–11 Search PubMed.
- A. Romero, M. Parentis, A. Habert and E. Gonzo, J. Mater. Sci., 2011, 46, 4701–4709 CrossRef CAS.
- D.-J. Liaw, K.-L. Wang, Y.-C. Huang, K.-R. Lee, J.-Y. Lai and C.-S. Ha, Prog. Polym. Sci., 2012, 37, 907–974 CrossRef CAS PubMed.
- T. Akhter, S. Saeed, H. M. Siddiqi and O. Ok Park, Polym. Adv. Technol., 2013, 24, 407–414 CrossRef CAS.
- T. Akhter, H. Siddiqi, S. Saeed, O. O. Park and S. Mun, J. Polym. Res., 2014, 21, 1–10 Search PubMed.
- X. Wang, L. Wang, Q. Su and J. Zheng, Compos. Sci. Technol., 2013, 89, 52–60 CrossRef CAS PubMed.
- L. Zou, S. Roddecha and M. Anthamatten, Polymer, 2009, 50, 3136–3144 CrossRef CAS PubMed.
- P. Musto, G. Ragosta, G. Scarinzi and L. Mascia, Polymer, 2004, 45, 1697–1706 CrossRef CAS PubMed.
- M. Nandi, J. A. Conklin, L. Salvati and A. Sen, Chem. Mater., 1991, 3, 201–206 CrossRef CAS.
- P. Musto, M. Abbate, M. Lavorgna, G. Ragosta and G. Scarinzi, Polymer, 2006, 47, 6172–6186 CrossRef CAS PubMed.
- Y. Li, G. He, S. Wang, S. Yu, F. Pan, H. Wu and Z. Jiang, J. Mater. Chem. A, 2013, 1, 10058–10077 CAS.
- G. Kickelbick, Prog. Polym. Sci., 2003, 28, 83–114 CrossRef CAS.
- G. Ragosta, P. Musto, M. Abbate and G. Scarinzi, J. Appl. Polym. Sci., 2011, 121, 2168–2186 CrossRef CAS.
- A. Morikawa, H. Yamaguchi, M. Kakimoto and Y. Imai, Chem. Mater., 1994, 6, 913–917 CrossRef CAS.
- Z. Ahmad and J. E. Mark, Chem. Mater., 2001, 13, 3320–3330 CrossRef CAS.
- H. Zou, S. Wu and J. Shen, Chem. Rev., 2008, 108, 3893–3957 CrossRef CAS PubMed.
- Y. Deng, M. Wu, X. Liu and Y. Gu, Eur. Polym. J., 2010, 46, 2255–2260 CrossRef CAS PubMed.
- C. Kizilkaya, S. Karataş, N.-K. Apohan and A. Güngör, J. Appl. Polym. Sci., 2010, 115, 3256–3264 CrossRef CAS.
- S. Saeed, M. Khalil and Z. Ahmad, J. Macromol. Sci., Part A: Pure Appl.Chem., 2009, 46, 152–162 CrossRef CAS.
- S. Sinha Ray and M. Okamoto, Prog. Polym. Sci., 2003, 28, 1539–1641 CrossRef PubMed.
- C. J. Cornelius and E. Marand, J. Membr. Sci., 2002, 202, 97–118 CrossRef CAS.
- C. J. Cornelius and E. Marand, Polymer, 2002, 43, 2385–2400 CrossRef CAS.
- S. H. Wang, Z. Ahmad and J. E. Mark, Chem. Mater., 1994, 6, 943–946 CrossRef CAS.
- L. Mascia and A. Kioul, Polymer, 1995, 36, 3649–3659 CrossRef CAS.
- S. Al-Kandary, A. A. M. Ali and Z. Ahmad, J. Mater. Sci., 2006, 41, 2907–2914 CrossRef CAS PubMed.
- S. Al-Kandary, A. A. M. Ali and Z. Ahmad, J. Appl. Polym. Sci., 2005, 98, 2521–2531 CrossRef CAS.
- C. Joly, M. Smaihi, L. Porcar and R. D. Noble, Chem. Mater., 1999, 11, 2331–2338 CrossRef CAS.
- J. W. Gilman, C. L. Jackson, A. B. Morgan, R. Harris, E. Manias, E. P. Giannelis, M. Wuthenow, D. Hilton and S. H. Phillips, Chem. Mater., 2000, 12, 1866–1873 CrossRef CAS.
- M. Khalil, S. Saeed and Z. Ahmad, J. Appl. Polym. Sci., 2008, 107, 1257–1268 CrossRef CAS.
- B. V. Kotov, T. A. Gordina, V. S. Voishchev, O. V. Kolninov and A. N. Pravednikov, Polym. Sci. U.S.S.R., 1977, 19, 711–716 CrossRef.
- M. Nishihara, L. Christiani, A. Staykov and K. Sasaki, J. Polym. Sci., Part B: Polym. Phys., 2014, 52, 293–298 CrossRef CAS.
- J. Ferraris, D. O. Cowan, V. Walatka and J. H. Perlstein, J. Am. Chem. Soc., 1973, 95, 948–949 CrossRef CAS.
- K. Balaji and S. C. Murugavel, J. Polym. Sci., Part A: Polym. Chem., 2011, 49, 4809–4819 CrossRef CAS.
- A. Tabbusum, Design, synthesis & characterization of some novel multifunctional molecules, Quaid-i-Azam University, Islamabad 45320, Pakistan, 2012 Search PubMed.
- W. Stober, A. Fink and E. Bohn, J. Colloid Interface Sci., 1968, 26, 62–69 CrossRef.
- Y. Gao, N. R. Choudhury, N. Dutta, J. Matisons, M. Reading and L. Delmotte, Chem. Mater., 2001, 13, 3644–3652 CrossRef CAS.
- V. Moravetski, J. R. Hill, U. Eichler, A. K. Cheetham and J. Sauer, J. Am. Chem. Soc., 1996, 118, 13015–13020 CrossRef CAS.
- D. H. Gray, S. Hu, E. Juang and D. L. Gin, Adv. Mater., 1997, 9, 731–736 CrossRef CAS.
- C. J. Chen, H. T. Lu, W. Y. Tseng, I. H. Tseng, S. L. Huang and M. H. Tsai, J. Appl. Polym. Sci., 2011, 122, 648–656 CrossRef CAS.
- Y. Chen and J. O. Iroh, Chem. Mater., 1999, 11, 1218–1222 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |