
Density functional study on the effect of a new ladder-type structure with different substituent groups (R = H, CH3, OCH3 and CN) for donor–acceptor copolymers†
Abstract
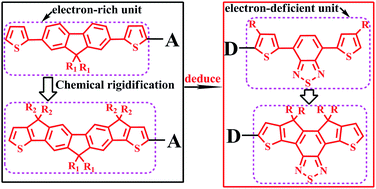
A series of ladder-type fused-ring D–A (donor–acceptor) copolymers have been designed and synthesized as electron donors for BHJ photovoltaic cells, in previous work, and almost all of them focus on electron-rich units. The aim of this paper is to explore the effect of a new ladder-type multifused electron-deficient unit (DCDTBT) with different substituent groups (R = H, CH3, OCH3 and CN) on the ground state structure, electronic, optical and charge transfer properties of D–A copolymers for the improvements of photovoltaic performance. Based on the reported D–A copolymer (P1) which was composed of an electron-rich benzo[1,2-b:4,5-b′]dithiophene unit (BDT) and an electron-deficient 4,7-di(4-hexyl-2-thienyl)-2,1,3-benzothiadiazole unit (4DTBT), the 4DTBT unit was replaced with the ladder-type di[4,4-dicyclopenta]dithienylbenzothiadiazole (DCDTBT) unit to form a new copolymer (P2). The calculated results reveal that P2 has better planarity, wider and stronger optical absorption, and smaller reorganization energy than P1. Then the substituent groups (CH3, OCH3 and CN) were introduced at the bridging carbons of the DCDTBT unit to design seven new donor copolymers (P2a–P2c, P2a′–P2c′ and P3). From the calculated results, the introduction of the substituent groups into the DCDTBT unit can markedly improve the electronic, optical and charge transfer properties of those copolymers, but have a little influence on molecular backbone planarity. In particular, the introduction of the cyano-group into the DCDTBT unit can obviously reduce the HOMO/LUMO levels of the copolymers. Interestingly, the copolymers (P2c/P2c′) combining the cyano-group and methyl or methoxyl into the same bridging carbon of DCDTBT unit have excellent electronic, optical properties and charge transfer ability, as promising donor polymers for high-efficiency (∼7% for P2c, ∼9% for P2c′) organic solar cells. In this work, a design strategy is used to select a suitable ladder-type electron-deficient unit for further improving the photovoltaic performance of donors in solar cells.
 Please wait while we load your content...
Please wait while we load your content...

