DOI:
10.1039/C4RA05903D
(Paper)
RSC Adv., 2014,
4, 32605-32621
Luminescence properties of Eu3+-activated SrWO4 nanophosphors-concentration and annealing effect†
Received
18th June 2014
, Accepted 4th July 2014
First published on 4th July 2014
Abstract
Eu3+ (2, 5, 7 and 10 at.%) activated SrWO4 phosphors were prepared via a polyol synthesis route at low temperature (∼150 °C) under urea hydrolysis. X-ray diffraction studies of all Eu3+ doped samples corroborate good crystallinity with a tetragonal scheelite-type structure of the SrWO4 phase. The value of strain was obtained in the range ∼0.003 to 0.004 for as-prepared (ASP) and samples annealed at 900 °C. Vibrational modes were studied using IR and Raman Spectroscopy. Photoluminescence studies of Eu3+ doped SrWO4 samples were carried out under 266, 394 and 464 nm excitations. An intense red emission was observed with a strong peak at ∼613 nm due to 5D0 → 7F2 transition under 266 nm. Effect of annealing at 900 °C on the photoluminescence properties of samples has been studied and it was found that luminescence intensity increases up to ∼9 times (for 10 at.% Eu3+-doped SrWO4) on annealing as compared to the corresponding as-prepared sample. High asymmetric ratios (A21) ∼ 6–13 were observed demonstrating it to be a red emitter. Calculated CIE co-ordinates of these Eu3+ doped samples under 266 nm excitation for ASP and annealed at 900 °C are x = 0.65 & y = 0.34, which are closer to the standard of NTSC (x = 0.67 & y = 0.33). These studies reveal that SrWO4:Eu3+ nano-phosphors can be used as potential red emitting phosphors for the development of white LEDs.
Introduction
Rare earth ion doped nano-materials have been extensively studied for their optical properties.1–4 Nano-materials are used as high-performance luminescent devices, catalysts, time-resolved fluorescence labels for biological detection and other functional materials based on their optical, electronic and chemical properties. Among others, special attention has been given to Eu3+ doped hosts emitting in the red region of the visible spectrum upon UV excitation due to their application in white LEDs.1–10 There is a need for red phosphors that have narrow line emission in deeper red regions for use in phosphor blends to produce fluorescent lamps. Due to its intense 5D0 → 7F2 emission in the red spectral region, Eu3+ has been extensively studied as an activator ion and used in most commercial red phosphors. Eu3+ ions doped tungstates have received considerable attention due to the special properties of WO42− group, such as high chemical stability, high X-ray absorption coefficient, and high average refractive index, which present efficient energy transfer from the tungstates host matrix to the localized states of the doping ions, and thus results in higher emission.11 Consequently, Eu3+-doped tungstate materials may serve as efficient red phosphors and would lead to the generation of highly efficient luminescent materials. Earlier investigations5,8,12,13 have showed that Eu3+ doped molybdates exhibited relatively strong absorption in the near-UV region and intense red emission with good color purity. Eu3+-doped tungstates may be the promising candidates as red emitting phosphors for the white LEDs applications.2,9 Various methods such as hydrothermal reaction,14,15 solid state reaction,2,9,11,16 co-precipitation method17 and sol–gel techniques3 have been employed in order to synthesize the Ln3+ doped and/or metal tungstates. For instance, Pereira et al.3 prepared SrWO4:Eu3+ by a non-hydrolytic sol–gel process and studied the effect of annealing temperature on its luminescent properties. Kang et al.9 synthesized MWO4 (M = Ca, Sr, Ba):Eu3+ by solid state reaction, and investigated its luminescent properties. The luminescent characteristics of SrWO4:Eu3+ phosphor for white LED was investigated by Ju et al.2 However, less attention has been paid on the effect of doping concentration on the luminescent properties of SrWO4:Eu3+ phosphors.
The most common method used for the synthesis of these phosphors is solid state method which requires heating at high temperatures for a long time and subsequent grinding. It results in aggregation, irregular shapes and damage of phosphor surfaces leading to decrease of luminescence intensity.3 Therefore, a simple, efficient and economically viable method for preparing highly efficient phosphors is desirable. The synthesis method used in the present study is the polyol method, which is well suited for the preparation of nano-sized metal oxide particles of various shapes.18–20 In this methodology, synthesis temperature is low (∼150 °C) and ethylene glycol (EG) is used as a reaction medium. The use of poly alcohols, like ethylene glycol, as reducing agents for obtaining metallic nano-particles has some advantages, as the by-products obtained during this process are ketones or carboxylic acids, which can be easily removed from the reaction mixture, unlike the residues of other reducing agents such as sodium boro-hydride or other boranes.
In this paper, we report the preparation of SrWO4:Eu3+ nano-phosphors with different concentration of Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) by polyol method followed by annealing at 900 °C in order to increase crystallinity. We have also investigated the luminescence properties of SrWO4:Eu3+ phosphors with varying concentration of Eu3+ ions in the host lattice. Since the ability to withstand high temperature is a basic requirement for phosphors applied in LEDs, the PL properties of the as-prepared and annealed at 900 °C samples have been studied. The color coordinates of ASP samples vary from (0.29, 0.23) to (0.65, 0.34) with Eu3+ concentration under 266, 394 and 464 nm excitation, providing a large scale tunability of multicolored emission. These chromaticity coordinates were compared with that of commercial red phosphor Y2O2S:Eu3+, and the results show that the chromaticity coordinates of phosphors synthesized in the present study are closer to the standard of NTSC (x = 0.67, y = 0.33) than the commercial red phosphor Y2O2S:Eu3+ (0.622, 0.351). In addition, the spectral distribution of SrWO4:Eu3+, as exhibited by its symmetrical emission peak, offers significant improvements over current red phosphors such as Y2O2S:Eu3+ or GdAlO3:Eu3+ (GAL), which have maintenance and stability concerns.21 Our results show that Eu3+ doped SrWO4 phosphor can be used as a potential red component for white light-emitting diodes.
Experimental details
Material synthesis
Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 were prepared using ethylene glycol (EG) as capping agent as well as reaction medium at 150 °C. Strontium nitrate (Sr(NO3)2, AR grade), europium oxide (Eu2O3, 99.99%, Sigma Aldrich) and sodium tungstate dihydrate (Na2WO4·2H2O, AR grade) were used as sources of Sr2+, Eu3+ and WO42−, respectively. In a typical synthesis procedure of 2 at.% Eu3+-doped SrWO4 nano-particles, 1.150 g of Sr(NO3)2 and 0.019 g of Eu2O3 were dissolved together in concentrated nitric acid (HNO3). The mixture was heated at 80 °C to remove the excess of acid and the process of removal of excess of acid was repeated five times after addition of de-ionized water (5 ml). To this solution, 1.829 g of Na2WO4·2H2O was added followed by 50 ml of EG. The pH of the solution was adjusted to 9–9.5. The resulting solution was then stirred for 1 hour. This solution is then transferred to a two neck round bottom flask and was heated up to 150 °C for 3 hours under refluxing condition in a condenser until precipitation was complete. The white precipitate so obtained was collected by centrifugation and washed 5 times in methanol to remove excess of EG and finally it was washed with acetone and dried at 90 °C for 2 hours in vacuum oven to yield the final white product. To obtain the optimal doping concentration of Eu3+ ions, SrWO4 phosphors doped with the concentrations of 2, 5, 7 and 10 atomic percentages (at.%) were synthesized by the same procedure, respectively. In this preparation method urea is used as a source of ammonia, which is a precipitating agent. Its decomposition temperature in water medium (120 °C) is lower than that in the gas/solid phase (190 °C) and it produces ammonia and CO2 or CNOH. Ammonia molecules react with Sr2+ and WO42− ions to form their oxides or/and hydroxides. During the reaction, ammonia also oxidizes WO32− to WO42− and nucleation of SrWO4 takes place. When the nucleation starts, surrounding EG molecules cap smaller particles and thus, particle growth is hindered. Also, the final dielectric medium does not allow further particle growth. Finally, the as prepared samples were divided in 2 parts. One part of the sample was annealed at 900 °C in an ambient atmosphere at a heating rate of 2 °C min−1 for 2 hours in an alumina crucible and the other part was left untreated.
Characterizations
The structures of the final products were characterized by using D8 Bruker X-ray diffractometer (XRD) with Ni-filtered Cu-Kα (1.5405 Å) radiation at 40 kV and 40 mA. All patterns were recorded over the range 10° ≤ 2θ ≤ 80° with a step size of 0.035°. The surface morphology and particle size of the nano-particles were characterized by Transmission Electron Microscopy (TEM) using a Tecnai G2 20 at an acceleration voltage of 200 kV. For TEM measurement, the samples were dispersed in methanol. A drop of the dispersed particles was put over the carbon coated copper grid and dried in the ambient atmosphere. Simultaneous DSC/TGA spectra were recorded using NETZSCH STA 449 F1. DSC and TG analyses were carried out using 10 mg of the sample at a heating rate of 10 °C min−1 up to 1000 °C, in nitrogen air under a flow of 60 cm3 min−1. Infrared spectra were recorded on a Fourier transform infrared (FT-IR) spectrophotometer of Shimadzu (model 8400 S) with a resolution of 2 cm−1 and in the range 400–4000 cm−1. For IR measurement the samples were mixed with KBr (Sigma Aldrich, 99.99%) in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5 ratio and this mixture was placed in a sample holder and the spectra were recorded. UV-vis spectra were recorded using UV-2700 Double beam spectrophotometer in the reflection mode. For UV-vis measurement the samples were dispersed in the methanol and then the spectra were recorded. The Raman spectra of the as prepared and annealed samples were recorded with Renishaw micro-Raman spectrometer attached with 633 nm laser as an excitation source. Photoluminescence measurements were carried out under ultraviolet excitation using 266 nm radiation from a Nd:YAG laser and detected by a CCD detector (Model: QE 65000, Ocean Optics, USA) attached with the fiber. Lifetime decay was recorded with Edinburg instrument F-920 equipped with 100 W μs flash xenon lamp as the excitation source.
:5 ratio and this mixture was placed in a sample holder and the spectra were recorded. UV-vis spectra were recorded using UV-2700 Double beam spectrophotometer in the reflection mode. For UV-vis measurement the samples were dispersed in the methanol and then the spectra were recorded. The Raman spectra of the as prepared and annealed samples were recorded with Renishaw micro-Raman spectrometer attached with 633 nm laser as an excitation source. Photoluminescence measurements were carried out under ultraviolet excitation using 266 nm radiation from a Nd:YAG laser and detected by a CCD detector (Model: QE 65000, Ocean Optics, USA) attached with the fiber. Lifetime decay was recorded with Edinburg instrument F-920 equipped with 100 W μs flash xenon lamp as the excitation source.
Results and discussion
XRD analysis
Fig. 1(a) and (b) show the XRD patterns of ASP and 900 °C annealed samples of Eu3+ (2, 5, 7 and 10 at.%) doped SrWO4, respectively. It can be seen from the figure that even the ASP samples are highly crystalline with tetragonal structure. All diffraction peaks match well with JCPDS card no. 08:0490 (a = 5.4168 Å, c = 11.951 Å and V = 350.66 Å3) of pure SrWO4. The absence of the peaks assigned to europium oxide (Eu2O3) and slight shift in the diffraction peaks to higher angles with respect to JCPDS card no. 08:0490 suggest the successful substitution of Sr2+ (1.18 Å) sites by Eu3+ (0.947 Å) ions, (Fig. 1(a)). However, some extra peaks with very small intensities (marked as #), are observed in case of Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 samples annealed at 900 °C, at an angle 2θ = 16.66 and 28.6°. These extra peaks are not matching with JCPDS card no. 08:0490 and their intensity increase with the Eu3+ ion doping concentration. These peaks may be related to WOn·mH2O (where m and n are whole numbers), other Eu3+ oxides, carbonates, Eu–W–O related compounds present on the surface of the samples but these peaks could not be distinguished in this study. Similar results have been reported for Gd3+ (2, 5, 7 and 10 at.%) co-doped CaMoO4:Eu3+ samples.22
 |
| | Fig. 1 XRD patterns (JCPDS no. 08-0490) of (a) as-prepared and (b) annealed at 900 °C SrWO4:Eu3+ nanophosphors with different concentrations of Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%). The symbol # shows the extra peaks evolution. | |
The lattice parameters of ASP 7 at.% Eu3+-doped SrWO4 are a = 5.413 Å, c = 11.927 Å, V = 349.60 Å3 and 900 °C annealed samples are a = 5.429 Å, c = 11.975 Å, V = 353.01 Å3. The diffraction patterns intensity of 7 and 10 at.% Eu3+-doped SrWO4 was found to be slightly less than 2 and 5 at.% doped samples which may be due to defects created at higher concentration in Eu3+-doped SrWO4 (Fig. 1(a)). Samples annealed at 900 °C show slightly better crystallinity than the ASP samples, which is shown in Fig. 1(b). However, with the increase in Eu3+ ion concentration the intensities of diffraction peaks decrease slightly, which indicates the defects associated with the sample surface and lattice distortion which has been also confirmed in PL study (discussed later).
The average crystallite size of the samples was calculated by the Scherrer formula,
| |
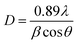 | (1) |
where,
D is the average crystallite size,
λ is the wavelength of the X-rays (0.15405 nm), and
θ and
β are the diffraction angle and full-width at half maximum (FWHM) of the peak in the XRD pattern, respectively. For
β correction,
βinst (FWHM due to instrument) is removed using Si standard. The strongest four peaks (1 1 2) at 2
θ = 27.653°, (2 0 0) at 2
θ = 33.063°, (2 0 4) at 2
θ = 45.136° and (3 1 2) at 2
θ = 55.803° were used to calculate the average crystallite size (
D) of the powders.
Fig. 2(a) and (b) show the variation of lattice parameters
a (Å),
c (Å) and unit cell volume, respectively with Eu
3+ ion concentration. In general, solid solutions show linear trends of lattice parameter in accordance with the ionic radius of substituted ion and its concentration.
23 It can be seen that the lattice parameters and cell volume decrease as Eu
3+ concentration increases from 2 to 10 at.%. This is due to the substitution of Sr
2+ (1.18 Å) ions by Eu
3+ (0.947 Å) ions.
24 Fig. 2(c) shows the variation of crystallite size with Eu
3+ ion concentration. Increase in crystallite size with increasing Eu
3+ concentration attributed to the increase in strain introduced due to the replacement of Sr
2+ ions by Eu
3+ ions of smaller radii. The unit-cell constants and the calculated average crystallite sizes of the ASP and 900 °C annealed samples of SrWO
4 with different concentrations of Eu
3+ ions are summarized in
Table 1.
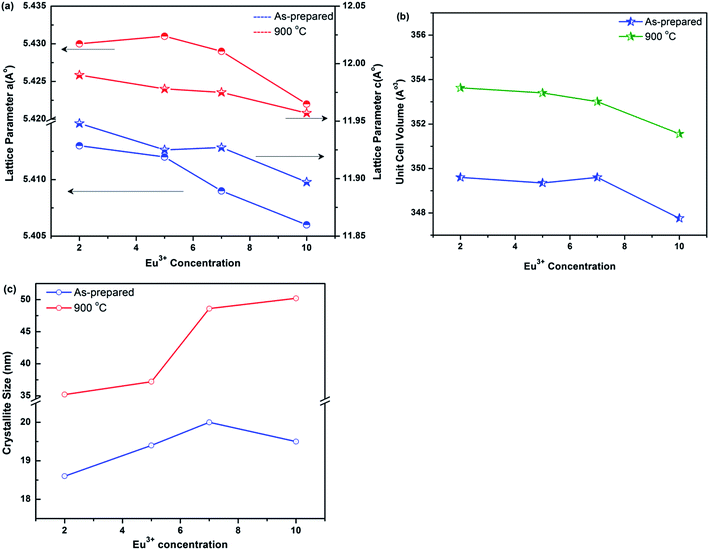 |
| | Fig. 2 (a) Lattice parameters, (b) unit cell volume and (c) crystallite size as a function of Eu3+ ions concentration. | |
Table 1 Unit cell constants, calculated average crystallite size and the strain of as-prepared and annealed at 900 °C SrWO4:Eu3+ nanoparticles
| Samples |
Eu3+ (at.%) |
Cell parameters |
c/a |
Cell volume (Å3) |
Crystal size (nm) |
Strain |
| a = b (Å) |
c (Å) |
| JCPDS 08-0490 |
|
5.416 |
11.951 |
2.206 |
350.66 |
|
|
| As-prepared |
2 |
5.409 |
11.948 |
2.208 |
349.60 |
18.6 |
0.00322 |
| 5 |
5.412 |
11.925 |
2.203 |
349.35 |
19.4 |
0.00380 |
| 7 |
5.413 |
11.927 |
2.203 |
349.60 |
20.0 |
0.00300 |
| 10 |
5.406 |
11.897 |
2.200 |
347.76 |
19.5 |
0.00345 |
| 900 °C |
2 |
5.430 |
11.990 |
2.208 |
353.63 |
35.2 |
0.00375 |
| 5 |
5.431 |
11.978 |
2.205 |
353.40 |
37.2 |
0.00392 |
| 7 |
5.429 |
11.975 |
2.205 |
353.01 |
48.6 |
0.00335 |
| 10 |
5.422 |
11.957 |
2.205 |
351.56 |
50.2 |
0.00395 |
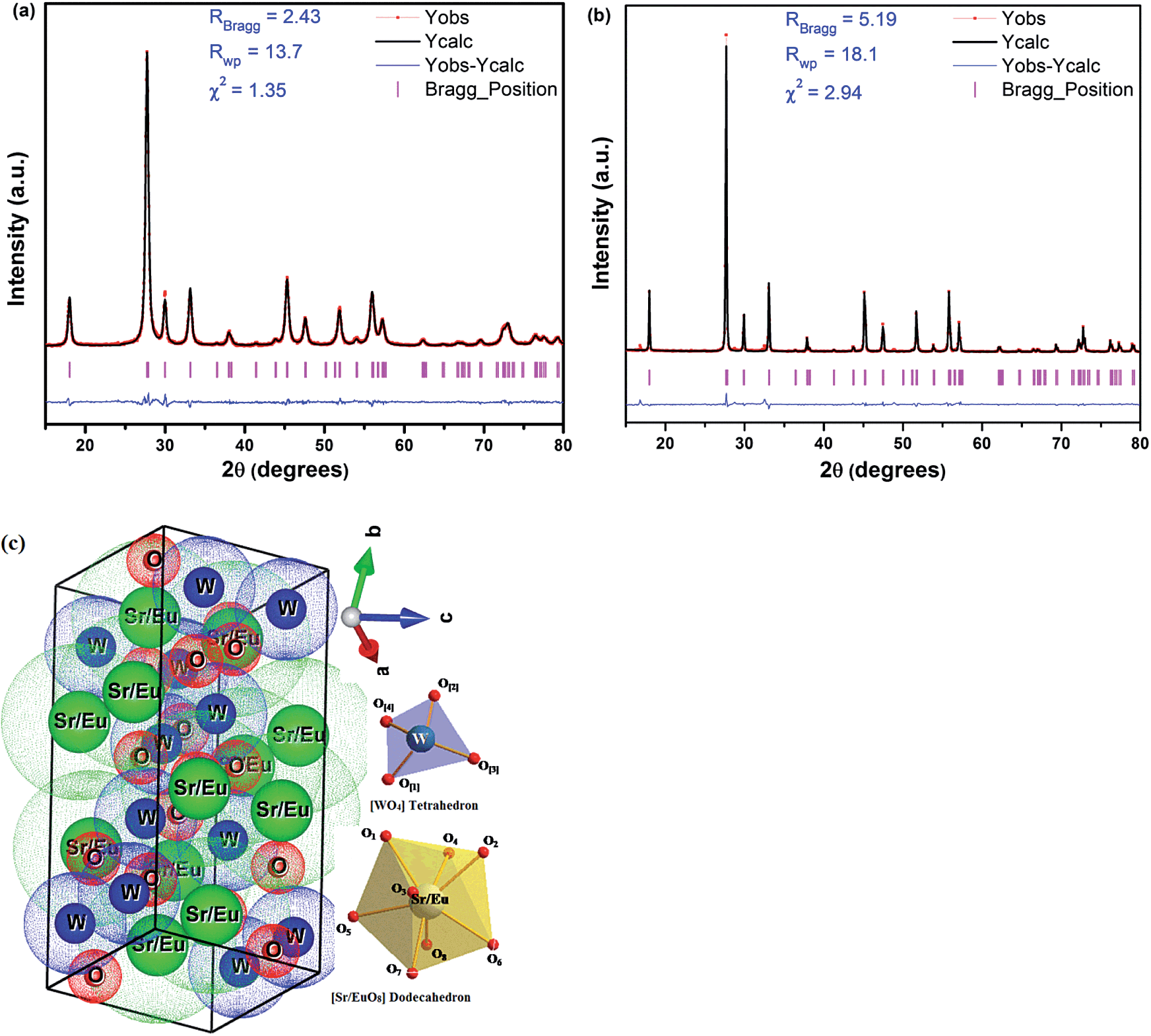
The strain induced in powders due to crystal imperfection and distortion was calculated using W–H (Williamson–Hall) fitting method.25 Strain ε was estimated from the slope of the linear fit to the data. The linear fit to the data for ASP 10 at.% Eu3+-doped SrWO4 is shown in Fig. 3. The value of strain was found in the range ∼0.0030 to 0.0040 for ASP and annealed samples (Table 1). Positive slope values corroborate the presence of tensile strain in the system. Similar behavior is reported in literature.22 The Rietveld analysis was performed for ASP and 900 ºC annealed 2 at.% Eu3+ doped SrWO4 samples (Fig. 4(a) and (b)) using the FullProf software.26 In the scheelite type tetragonal structure, positions of the respective atoms are Sr: (4b: 0, 0.25, 0.0625), W: (4a: 0, 0.025, 0.125) and O: (16f: x, y, z) with angles (α = β = γ = 90°). Pseudo-Voigt function was used to model the peak profiles and six coefficient polynomial was used to describe the background. (Yobs − Ycalc), Rwp, RBragg and χ2 are shown in figure itself (Fig. 4(a) and (b)). Rietveld plots of 5, 7 and 10 at.% Eu3+ doped SrWO4 nano-phosphors are shown in (ESI) Fig. S1(a)–(f).† Table 2 shows the atomic co-ordinates for Sr, W, O and Eu atoms obtained after the Rietveld refinement of 2 at.% as-prepared and annealed at 900 °C SrWO4:Eu3+ nano-particles. Fig. 4(c) illustrates the schematic representation of SrWO4:Eu tetragonal unit cell with a I41/a space group which was modeled by using the VESTA Software, which shows the presence of high inversion symmetry in the lattice. In Fig. 4(c) Sr2+ and Eu3+ cations are coordinated to eight oxygen atoms considered as [SrO8] and [EuO8] groups which form a distorted dodecahedron geometry. W atoms are coordinated to four oxygen atoms and form [WO4] groups with a slight distortion in the tetrahedral geometry.
 |
| | Fig. 3 Plot of βcosθ/λ vs. sinθ/λ of 10 at.% Eu3+-doped SrWO4 sample. | |
 |
| | Fig. 4 Rietveld plots of 2 at.% Eu3+ doped SrWO4 samples (a) as-prepared and (b) annealed at 900 °C and (c) simplified polyhedral representation of SrWO4:Eu showing both [Sr/EuO8] and [WO4] clusters. | |
Table 2 Wyckoff positions for Sr, W, O and Eu atoms obtained after the Rietveld refinement of 2 at.% as-prepared and 900 °C annealed SrWO4:Eu3+
| Atom |
Site |
SrWO4:2Eu3+ (ASP) |
SrWO4:2Eu3+ (900 °C) |
| x |
y |
z |
x |
y |
z |
| Sr |
4b |
0 |
0.25 |
0.625 |
0 |
0.25 |
0.625 |
| W |
4a |
0 |
0.25 |
0.125 |
0 |
0.25 |
0.125 |
| O |
16f |
0.2648 |
0.1141 |
0.0475 |
0.2441 |
0.6088 |
0.0395 |
| Eu |
4b |
0 |
0.25 |
0.625 |
0 |
0.25 |
0.625 |
Table 3 CIE values for different concentrations of Eu3+ doped SrWO4 phosphors under 266, 394 and 464 nm excitation
| Excitation (nm) |
Eu3+ (at.%) |
CIE coordinates |
| As-prepared |
Annealed (900 °C) |
| No. |
X |
Y |
No. |
X |
Y |
| 266 nm |
2 |
a1 |
0.65 |
0.34 |
x1 |
0.65 |
0.34 |
| 5 |
a2 |
0.63 |
0.34 |
x2 |
0.65 |
0.34 |
| 7 |
a3 |
0.58 |
0.35 |
x3 |
0.64 |
0.33 |
| 10 |
a4 |
0.58 |
0.35 |
x4 |
0.63 |
0.34 |
| 394 nm |
2 |
b1 |
0.46 |
0.28 |
y1 |
0.43 |
0.25 |
| 5 |
b2 |
0.36 |
0.24 |
y2 |
0.43 |
0.25 |
| 7 |
b3 |
0.34 |
0.24 |
y3 |
0.34 |
0.23 |
| 10 |
b4 |
0.29 |
0.23 |
y4 |
0.37 |
0.26 |
| 464 nm |
2 |
c1 |
0.48 |
0.36 |
z1 |
0.47 |
0.38 |
| 5 |
c2 |
0.40 |
0.38 |
z2 |
0.49 |
0.37 |
| 7 |
c3 |
0.35 |
0.39 |
z3 |
0.37 |
0.38 |
| 10 |
c4 |
0.31 |
0.40 |
z4 |
0.46 |
0.37 |
TEM study
Fig. 5(a) illustrates the TEM micrograph and corresponding SAED pattern of 5 at.% Eu3+-doped SrWO4 nano-particles annealed at 900 °C. Most of these SrWO4:Eu3+ nano-particles have irregular shapes and a few have the spherical shape. It is well known that the uniformity of the size and shape is controlled by nucleation.27 In the present preparation method, reaction was started after adding ethylene glycol. During the heating process, nucleation and crystal growth continued. This may be the reason for irregular shapes and agglomerated particles. The particle sizes determined from TEM is around 35–52 nm, which is in agreement with the calculated sizes from XRD studies. SAED studies are done on relatively isolated particles and resulted patterns are shown in Fig. 5(b). The SAED pattern in Fig. 5(b) shows the sharp diffraction spots corresponding to d-spacing values (020), (200) and (220) planes, which are the characteristics planes for scheelite type tetragonal SrWO4. The SAED pattern along the [001] zone axis exhibit isolated spots, where its distances denote a typical pattern of scheelite structure.
 |
| | Fig. 5 Images of (a) TEM and (b) corresponding SAED pattern of 5 at.% Eu3+ doped SrWO4 nanoparticles annealed at 900 °C. | |
DSC-TGA study
To study the thermal stability of prepared nano-phosphors, TGA (Thermo Gravimetric Analysis) was carried out. Fig. 6 shows the simultaneous DSC/TGA curves along with DTA (Differential Thermo Gravimetric Analysis) curves of ASP 2 at.% Eu3+ doped SrWO4. The sample is measured in the temperature range of 35 to 1000 °C with a heating rate of 10 °C min−1 under nitrogen atmosphere. The mass loss before 100 °C in the TG curve is due to the evaporation of adsorbed water. Nearly ∼5.5% weight loss has been observed up to 600 °C, while no significant weight loss has been monitored beyond 600 °C, which suggests that up to 600 °C, decomposition of all organic constituent precursors such as ethylene glycol had been carried out. There is an exothermic peak in the vicinity of 100 °C in the DTA curve which corresponds the evaporation of water molecules in the sample.
 |
| | Fig. 6 Simultaneous DSC/TG character of as-prepared 2 at.% Eu3+-doped SrWO4. | |
To study the heat flow as a function of temperature in the inert gas (N2) atmosphere associated with transitions in SrWO4:2Eu3+ ASP sample DSC was recorded (Fig. 6). The curve shows both exothermic and endothermic peaks. The peak around 143 °C represents the mass loss due to evaporation of water and methanol. However, the exothermic peak centered about 231.9 °C, signifies the evaporation of organic compounds in the sample. Whereas, the large and sharp endothermic DSC (Differential Scanning Calorimetry) peak at about 674 °C indicates that the phase formation of SrWO4:2Eu3+ starts around this temperature. These results show that the prepared nano-phosphors are thermally stable and can be used in lightning and display devices.
FTIR study
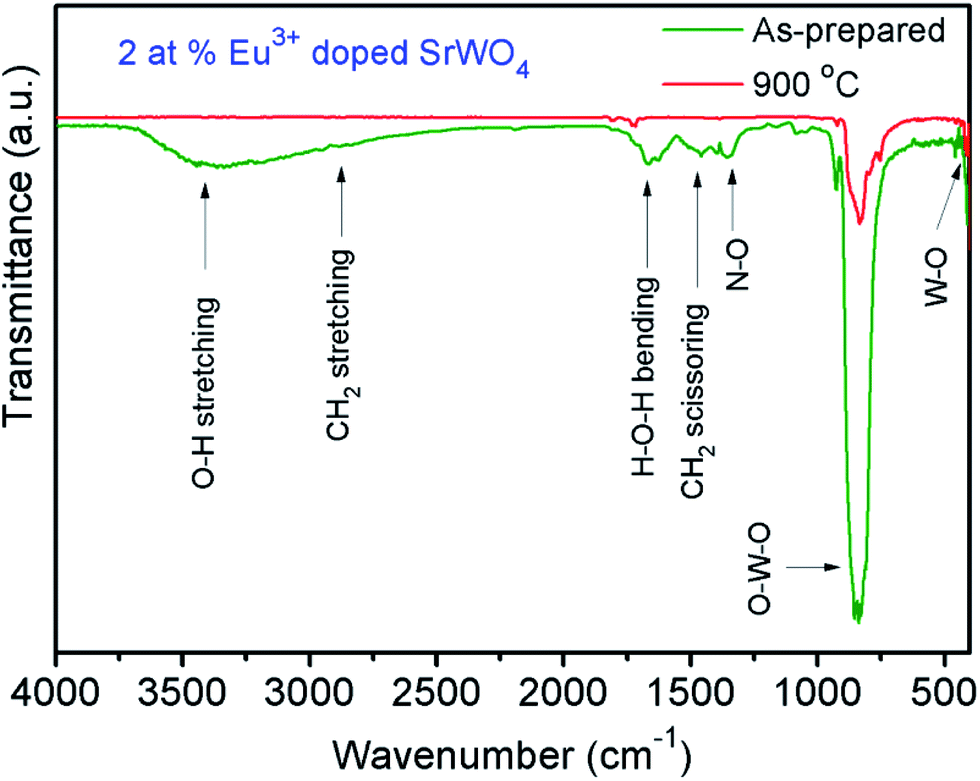
FT-IR spectra of the ASP and 900 °C annealed nano-particles of 2 at.% Eu3+-doped SrWO4 in the wave-number range of 400–4000 cm−1 are shown in Fig. 7. Different vibrational modes observed for ASP SrWO4:Eu3+ are listed in Table S1.†
 |
| | Fig. 7 FTIR spectra of as-prepared and 900 °C annealed 2 at.% Eu3+-doped SrWO4 samples. | |
The bands at 1666 and 3444 cm−1 correspond to H–O–H bending and O–H stretching vibrations of water molecules present on the surface of nano-particles, respectively.28 The absorption intensities of these peaks decrease on annealing the samples at 900 °C which indicates the evaporation of adsorbed water from the sample surfaces (Fig. 7). The absorption peaks appearing in the range of 400–1000 cm−1 are the typical FT-IR absorption peaks of WO42− with scheelite tetragonal structure.2,14 The band at 837 cm−1 is due to the O–W–O asymmetric stretching vibration of the WO42− tetrahedron and 457 cm−1 corresponds to the bending vibration of W–O (Au mode). It is found that both the O–W–O stretching vibration and W–O bending vibration are shifted slightly to lower wave-number by ∼3–5 cm−1 (i.e., red shift) on annealing the samples at 900 °C. The red shift of Au is responsible for lattice expansion of SrWO4. Similar behavior was observed by Su et al.15 in the case of CaWO4 nano-particles. The FWHM value at 837 cm−1 decreases with heat treatment. Modes of vibration at 1357 cm−1 are due to the N–O band from HNO3 used in the sample preparation. In the ASP sample peaks are observed near 2946 cm−1 indicating C–H stretching vibration from EG molecules on the surface of SrWO4:Eu3+ nano-particles whereas, its bending vibration (scissoring) is obtained at 1456 cm−1. The prepared nano-particles are able to disperse in polar compounds such as water and methanol, due to the presence of hydrogen bonding.1
Raman study
Fig. 8 shows Raman spectra at 633 nm excitation in the frequency ranging from 100 to 1100 cm−1 for SrWO4:Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) nano-particles annealed at 900 °C. At room temperature all samples show well known Raman bands at ∼104, 131, 188, 334, 371, 797, 836 and 920 cm−1 and weak bands near 264 and 405 cm−1. Stronger Raman-active vibrational modes indicate a strong interaction between ions which is mainly due to stretching and bending vibrations of metal–oxygen bonds within anionic groups.17 In the present study, we have studied the vibrations of W–O within WO42−. In comparison to metal–metal vibrational modes Raman spectroscopy is more sensitive to metal–oxygen vibrational modes.29
 |
| | Fig. 8 Room temperature Raman spectra of SrWO4:xEu3+ nanopowders (a) x = 2 at.% (b) x = 5 at.% (c) x = 7 at.% and (d) x = 10 at.% processed by the polyol method and treated at 900 °C for 2 h. | |
The Raman peak at 920 cm−1 could be assigned as ν1 of the O–W–O symmetric stretching while the peak at 334 cm−1 is assigned as ν2 of the W–O symmetric bending. The peaks at 836 and 797 cm−1 are designated as ν3 of the O–W–O anti-symmetric stretching, and peaks at 371 and 405 cm−1 are designated as ν4 of the W–O anti-symmetric bending.30,31 External peak modes are localized at a range from 104 to 131 cm−1 which corresponds to the stretching and flexion mode of Sr–O. Free rotation modes of WO42− are visible at 188 and 264 cm−1. Vibrational modes are in accordance with analyzed Raman vibrations. According to literature data,32 all Raman modes observed for SrWO4:Eu3+ obtained in this work are characteristics of the tetragonal structure. The compounds annealed at 900 °C with doping concentration of 2 and 5 at.% (Fig. 8(a) and (b)) show some strong peaks of ν3 at 836 and 797 cm−1 which are related to internal modes; peak characteristics of νs.r are visible at 104 cm−1 which are external modes.
Raman modes with lower intensities are also observed due to local disorder and defects present in the scheelite lattice of SrWO4. Similar results have been reported29 in case of pyrochlore lattice of EYYTO. Raman spectra for these samples with different concentrations of Eu3+ did not represent any significant change which indicates that the doping concentration of europium was unable to modify stretching, torsion and bending vibrational modes of W–O bonds since the W atom is located at the B site of the scheelite structure, while Eu3+ ion replaces the Sr2+ ion located at the A site in the SrWO4 matrix.3 The Raman spectra at 514 nm of ASP and 900 °C annealed samples of 7 at.% Eu3+-doped SrWO4 are shown in Fig. S3 (ESI†), which also show the similar pattern as shown by Raman spectra at 633 nm.
UV-vis spectroscopy
Fig. 9 shows the UV-vis absorption spectra of Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 nano-particles annealed at 900 °C, exhibiting strong absorption band edge at 285, 275, 265 and 261 nm, respectively. These bands are attributed to a charge transfer from the oxygen ligands to the central tungstate atom within the WO42− group ions. Weaker bands originating from the Eu3+ f–f transitions were also observed in the region 350–400 nm. This (narrower absorption peak in UV region) is in good agreement with the formation of nano-sized strontium tungstate particles with a narrow particle size distribution.33,34
 |
| | Fig. 9 UV-vis absorption spectra for the SrWO4:Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) nanoparticles annealed at 900 °C. | |
The optical band gap energy was estimated by the method proposed by Wood and Tauc.35 According to them the optical band gap is associated with the absorbance and incident photon energy by the following equation:
where,
k is a constant,
h is the Planck's constant,
ν is the frequency,
Eg is the optical band gap and exponent
n may have the values 1/2, 2, 3/2 or 3 depending on whether the transition is direct allowed, indirect allowed, direct forbidden or indirect forbidden, respectively. It is reported
36–38 that tungstates have an optical absorption governed by direct electronic transitions due to its straight line behavior in the high energy region. The band gap energy
Eg can be obtained from an extrapolation of the straight-line portion of the (
αhν)
2 versus hν plot to zero absorption coefficient value.
Fig. 10(a) and (b) provides the estimated band gap 4.25 eV and 3.98 eV for ASP and 900 °C annealed 2 at.% Eu
3+-doped SrWO
4 nano-particles, respectively. The band gap calculations for 900 °C annealed SrWO
4:Eu
3+ (Eu
3+ = 5, 7 and 10 at.%) nano-particles are shown in ESI Fig. S2(a)–(c)
† and estimated to be ∼3.98, 3.96 and 3.99 eV. SrWO
4:Eu
3+ ASP nano-particles exhibit a typically continuous smooth absorption that increases as a function of the energy. The band gap energy clearly shifts to a lower energy on annealing the samples (
Fig. 10). This red shift in band gap energy can be associated to quantum confinement effect in nano-size particles
i.e. the squeezing of electron hole pair below the dimensions approaching exciton Bohr radius and with the intermediate localized states in the band gap due to structural defects.
39,40
 |
| | Fig. 10 Variation of (αhν)2 vs. photon energy (in eV) curve for (a) as-prepared and (b) 900 °C annealed 2 at.% Eu3+-doped SrWO4. | |
Fig. 11 shows the energy level diagram of Eu3+-doped SrWO4. On irradiating the samples under ∼266 nm (4.66 eV), the electrons get excited to W–O CT bands and by non-radiative transitions these electrons relax to lower energy states and then by transferring their energies they excite Eu3+ ions to higher energy states (5D0). These excited Eu3+ ions return to the ground states of Eu3+ (7F1, 7F2, 7F3 and 7F4) by radiative transition in red region.
 |
| | Fig. 11 Schematic energy level diagram for transfer process between WO42− and Eu3+ ions in SrWO4. | |
Photoluminescence study
Fig. 12(a) shows the excitation spectra of 900 °C annealed Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%)-doped SrWO4 nano-particles by monitoring the emission wavelength at 613 nm. A broad band from 230 to 320 nm and the f–f transitions of Eu3+ around 360 (7F0 → 5D4), 382 (7F0 → 5G2), 393 (7F0 → 5L6), 415 (7F0 → 5D3) and 464 (7F0 → 5D2) are clearly observed. These were assigned to the combination of the ligand to metal charge transfer (O2− → W6+) and charge transfer band (CTB) from the completely filled 2p orbitals of O2− to the partially filled f–f orbitals of the Eu3+ ions (O2− → Eu3+).2,3,16 The wavelength corresponding to the peak of Eu–O CT band around 230–320 nm decreases from 265 to 255 nm with the increase in Eu3+ ion concentration upto 10 at.% (Fig. S4 (ESI†)). This blue shift in the Eu–O CT band with Eu3+ ion concentration can be related to the change in the crystal field splitting of Eu3+ ions. It is in accordance with the reports by Robertson et al.41a and Jang et al.,41b they have expressed the crystal field splitting (Dq) as:| |
 | (3) |
where, Dq is a measure of energy level separation, Z is the charge on anion, e is the charge on an electron, r is the radius of the f wavefunction and R is the bond length. When Sr2+ site is substituted and occupied by smaller Eu3+ ion, the distance between Eu3+ and O2− becomes longer. Since crystal field splitting is proportional to 1/R5, this longer Eu3+ and O2− distance also decreases the magnitude of the crystal field, resulting smaller crystal field splitting, so that there is continuous increase in blue shift with increase in Eu3+ ion concentration.41c
 |
| | Fig. 12 (a) Excitation spectra of 900 °C annealed Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 nanoparticles under 613 nm excitation, (b) excitation spectra of 2 at.% Eu3+ doped SrWO4 nanoparticles annealed at 900 °C. Inset shows the resolution of dual peaks between 230 to 320 nm due to Eu–O and W–O CT and (c) excitation spectra (after excitation at 613 nm) of 10 at.% Eu3+ doped SrWO4 nanoparticles for as-prepared and 900 °C annealed samples. | |
Fig. 12(b) shows the excitation spectrum of 900 °C annealed 2 at.% Eu3+-doped SrWO4 sample monitored at 613 nm, it consists of strong absorption band due to Eu–O and W–O charge transfers (CT) between 230 to 320 nm (shown in the inset of Fig. 12(b)), which shows the two resolved peaks. The peaks calculated by fitting the data with a Gaussian distribution are assigned to Eu–O and W–O CT bands at 266 and 286 nm, respectively. In case of ASP samples these peaks could not be clearly distinguished (Fig. S4 (ESI†)). The absorption intensity of 7F0 → 5L6 transition at 394 nm (FWHM ∼ 5 nm) is 9.4 times stronger than Eu–O CT absorption which suggests a weak energy transfer from Eu–O CT band to Eu3+ (Fig. 12(b)).
Fig. 12(c) shows the excitation spectra of ASP and 900 °C annealed 10 at.% Eu3+-doped SrWO4 nano-phosphors at 613 nm emission wavelength. The peak maximum of broad peak (230–310 nm) arising due to Eu–O CT shifted from 270 nm (as-prepared) to 280 nm on annealing at 900 °C. It is in accordance with the earlier reported results.27 Luminescence intensity of 7F0 → 5L6 transition at 394 nm of 900 °C annealed SrWO4:10 Eu3+ sample increases by 15 times as compared to its ASP sample. This may be due to decrease in H2O molecules, non-radiative rates arising from OH group and EG molecules on the surface of the nano-particles.
Fig. S4 (ESI†) shows the excitation spectra of ASP SrWO4:Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) nano-particles monitored at 613 nm emission wavelength. It shows the same luminescence profile; however, there are some differences in peak positions and luminescence intensity. To observe PL intensity of SrWO4:Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) samples, luminescence spectra for ASP and 900 °C annealed samples were recorded at different excitation 266 nm (host or Eu–O CT), 394 nm (direct Eu3+) and 464 nm (direct Eu3+) wavelengths. Fig. 13(a) and (b) show the emission spectra of Eu3+-doped SrWO4 (Eu3+ = 2, 5, 7 and 10 at.%) for ASP and 900 °C annealed samples under the excitation at 266 nm with a Nd-YAG laser, respectively. Four characteristic emission peaks have been observed at 590, 613, 651 and 700 nm corresponding to 5D0 → 7F1, 5D0 → 7F2, 5D0 → 7F3 and 5D0 → 7F4 transitions of Eu3+, respectively. In the emission spectra the strongest peaks are observed at 613 nm and the samples show a predominant red emission of the characteristic Eu3+ (5D0 → 7F2) transition under 266 nm excitation. Due to magnetic dipole transition (5D0 → 7F1), orange-red emission lines originate around 590 nm while the 5D0 → 7F2 lines originate due to electric dipole transition. According to the Judd–Ofelt theory, the magnetic dipole transition is permitted. But when the europium ion occupies a site without an inversion centre and the intensity is significantly affected by the symmetry in local environments around Eu3+ ions, only electric dipole transition is allowed.3,10,42–46 The magnetic dipole transition 5D0 → 7F1 is the dominant transition only when Eu3+ ions occupy an inversion symmetry site in the SrWO4 crystal lattice, otherwise electric dipole transition 5D0 → 7F2 is dominant. It can be observed from the emission spectra that electric dipole transition is dominant. This suggests that most of the Eu3+ enters into the lattice sites having centre without inversion symmetry. From Fig. 13(a) and (b), it can be seen that the peak position of the emission lines are almost the same, but the intensity patterns are drastically different.
 |
| | Fig. 13 Emission spectra of (a) as-prepared and (b) 900 °C annealed Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 nanoparticles at 266 nm excitation and (c) Emission spectra (after excitation at 266 nm) of 10 at.% Eu3+ doped SrWO4 nanoparticles for as-prepared and 900 °C annealed samples. Inset shows the 5D1 → 7F1 transition of the Eu3+ at ∼530 nm. | |
Fig. 13(c) shows the emission spectra of ASP and 900 °C annealed 10 at.% Eu3+ doped SrWO4 sample. The excitation wavelength used was 266 nm. The peak intensities corresponding to Eu3+ are relatively low in the case of ASP sample. The luminescence intensity of Eu3+ in case of as-prepared 10 at.% Eu3+ doped SrWO4 under 613 nm peak increases up to ∼9 times on annealing the sample at 900 °C. This is due to the extent of reduction of non-radiative processes arising from –OH vibrations on the surface of the nano-particles, increase in particle size and also due to lowered surface to volume ratio after annealing. Particle size is small in case of as-prepared sample also the surface has H2O and EG molecules, which acts as luminescence quencher. But on annealing the samples at 900 °C, most of the carbon impurities and H2O are removed, EG gets decomposed and particle size increases as shown by XRD and TEM studies. This enhances the luminescence intensity. The position of W–O charge-transfer band (CTB) is shifted to higher wavelength by ∼2–5 nm due to annealing of the samples at 900 °C than in ASP samples; similar observations have been reported in Mo–O CTB.28 This can be explained as follows: when an electron is transferred from oxygen to W, an electronic transition takes place which gives rise to the W–O charge transfer band (CTB). In ASP samples, there are a relatively large number of dangling bonds over the particle surface and the lattice is less ordered as compared to that of annealed samples. In these spectra, the presence of the band around 530 nm was also observed; which is attributed to 5D1 → 7F1 transition of the Eu3+ (inset of Fig. 13(c)). This transition indicates that the Eu3+ ions are located in at least two different chemical surroundings in the SrWO4 lattice. On annealing the sample at higher temperature, the intensity of broad band at around 530 nm under 266 nm excitation decreases due to decrease of lattice defects and thus energy transfer from host WO42− to Eu3+ increases and Eu3+ emission intensity is enhanced.47
Fig. 14(a) and (b) and 15(a) and (b) show the emission spectra of Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 under 394 and 464 nm excitations, respectively. The luminescence intensity is also influenced by excitation wavelength.48 Annealing temperature will increase the fluctuations in emission intensity49 (Fig. 14(b) and 15(b)). The optimum concentration in case of as-prepared samples is 2 at.% while in case of 900 °C annealed samples it is 7 at.% (Fig. 14). This variation can be explained as:
 |
| | Fig. 14 Luminescence spectra of (a) as-prepared and (b) 900 °C annealed Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 nanoparticles at 394 nm excitation. | |
 |
| | Fig. 15 Emission spectra of (a) as-prepared and (b) 900 °C annealed Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 nanoparticles at 464 nm excitation. | |
Lanthanide doped systems have a typical property of showing sensitized emission up-to a certain doping concentration and beyond an optimum concentration, luminescence quenches. This is due to the decrease of mean distance between the neighboring lanthanide ions below the critical value, which leads to cross-relaxation among them and thus probability of radiative transition is reduced.50 On annealing at higher temperature due to increase of Eu3+ occupancy in the Sr2+ lattice sites, the extent of reduction of non-radiative rates and with the improvement of crystallinity optimum concentration increases.3 Our results are supported by the observations reported in literature.5,28,48–52 They have reported similar disorder in case of samples annealed at higher temperatures.
In order to study the change in luminescence intensity of 5D0 → 7F2 (613 nm) transition with Eu3+ ion concentration, variation of peak intensity (integrated area under the curve) with Eu3+ ion concentration has been observed (Fig. 16(a) and (b)). The curves are fitted and the areas under the emission peaks are calculated using Gaussian distribution function, which is represented as:
| |
 | (4) |
where,
I is the observed intensity,
I0 is the background intensity,
wi the FWHM of the curve,
Ai the area under the curve,
λ the wavelength and
λci the mean value corresponding to the transition. Before fitting the curve, the baseline correction was done (
I0 = 0). Two curves are fitted within the range 580–600 and 600–630 nm for
5D
0 →
7F
1 (590 nm) and
5D
0 →
7F
2 (613 nm) transitions, respectively. The variation of integrated emission intensity (A
2) of the
5D
0 →
7F
2 transition (613 nm) with Eu
3+ ion concentration (Eu
3+ = 2, 5, 7 and 10 at.%) at different excitation wavelength is shown in
Fig. 16(a) and (b). In case of ASP samples, intensity (A
2) decreases with increase in Eu
3+ ion concentration, which may be attributed to concentration quenching effect. The inset of
Fig. 16(a) shows the digital photograph of 2 at.% Eu
3+-doped SrWO
4 before and after irradiation under 266 nm laser excitation (Nd-YAG); an intense red color was observed. In case of 900 °C annealed samples luminescence intensity (A
2) decreases with Eu
3+ ion concentration up to 5 at.% and then increases with further increase in Eu
3+ (
Fig. 16(b)). The increase in luminescence with Eu
3+ shows substitution of Sr
2+ by Eu
3+. The change in FWHM (
w2) with Eu
3+ ion concentration (Eu
3+ = 2, 5, 7 and 10 at.%) for ASP and 900 °C annealed SrWO
4:Eu
3+ nano-phosphors at different excitations are shown in
Fig. 17(a) and (b), respectively. The FWHM (
w2) of
5D
0 →
7F
2 transition at 613 nm varies between ∼5–9 nm for ASP and 900 °C annealed samples. For ASP samples the FWHM (
w2) increases with increase in Eu
3+ ion concentration. FWHM values of annealed samples are smaller than the ASP samples. Smaller FWHM (
w2) means a sharper peak and usually high luminescence intensity. This corroborates with an increase in luminescence for annealed samples. It was observed that
w2 decreases with heat treatment.
5
 |
| | Fig. 16 Variation of integrated area of 5D0 → 7F2 transition (A2) for (a) ASP and (b) 900 °C annealed SrWO4:Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) nanophosphors at different excitations. Inset shows a photograph of 2 at.% Eu3+-doped SrWO4 before and after irradiation under 266 nm (Nd-YAG) laser excitation. | |
 |
| | Fig. 17 Variation of FWHM (w2) of transition 5D0 → 7F2 as a function of Eu3+ ion concentration (Eu3+ = 2, 5, 7 and 10 at.%) for (a) ASP and (b) 900 °C annealed SrWO4:Eu3+ nanophosphors at different excitations. | |
The ratio of integrated area of electric dipole transition (5D0 → 7F2) to magnetic dipole transition (5D0 → 7F1) of Eu3+ ion has been used to study the structural distortion around the Eu3+ ion, it is known as asymmetric ratio (A21).53,54 In our case the asymmetric ratio is represented as,
| |
 | (5) |
where, subscript ‘2’ and ‘1’ refer to transitions of
5D
0 →
7F
J,
J = 2 and 1, respectively. Lesser the value of A
21 ratio more symmetric is the system.
40 Fig. 18(a) and (b) show the asymmetric ratio (A
21 = ∫
5D
0 →
7F
2/∫
5D
0 →
7F
1) as a function of Eu
3+ ion concentration for ASP and 900 °C annealed samples of SrWO
4:Eu
3+, respectively. For ASP samples, A
21 values decreases with Eu
3+ ion concentration upto 7 at.% and lies between ∼7.9 to 13.2. A
21 ratio increases with increase in Eu
3+ ion concentration for 900 °C annealed samples and lies between ∼6.0 to 13.4. Overall, the A
21 values increase on annealing because of decrease of non-radiative rates and increase of asymmetry around Eu
3+ ion. Increase in A
21 values demonstrates high distortion present in the host lattice which shows high red emission.
49
 |
| | Fig. 18 The asymmetric ratio (A21 = ∫5D0 → 7F2/∫5D0 → 7F1) versus Eu3+ ion concentration for (a) ASP and (b) 900 °C annealed Eu3+ doped SrWO4 nanoparticles (Eu3+ = 2, 5, 7 and 10 at.%) at 266, 394 and 464 nm excitations. | |
The critical distance (Rc) between Eu–Eu ions is used to understand the energy transfer and concentration quenching, it can be calculated through the relation given by Blasse,55
| |
 | (6) |
where,
V is the unit cell volume,
Co is the optimum concentration (at.%) and
N is the number of dopant sites available in the unit cell. In our case, by taking
Co = 0.02,
N = 4 and
V = 349.65 Å
3, the critical distance
Rc are found to be ∼20.28 and 20.35 Å for ASP and 900 °C annealed 2 at.% Eu
3+-doped SrWO
4, respectively. When the distance between Eu–Eu,
REu–Eu <
Rc, energy transfer between Eu to Eu dominates, otherwise energy is transferred from WO
42− ions.
PL emission is attributed to the presence of defects into the band gap of crystalline SrWO4:Eu3+ powders, which favor the electronic transitions within the localized energy levels.56 The red emission in the SrWO4:Eu3+ nano-phosphors can be associated with the disorder in the long and short ranges and the distorted [WO4] tetrahedron due to the different angles between the O–W–O. It has been reported3,57 that the lattice disorder is associated to the presence of oxygen vacancies in disordered tungstates powders that occur in three different charge states: the  state, which becomes neutral by capturing electrons,
state, which becomes neutral by capturing electrons,  state, which is singly ionized and the doubly positively charged state in the lattice
state, which is singly ionized and the doubly positively charged state in the lattice 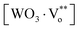 which does not trap any electrons. When a trivalent metallic ion such as Eu3+ is incorporated into a host lattice and substituted for a divalent metallic ion such as Sr2+, then it is difficult to keep a charge balance. In order to maintain the charge balance the host has to capture O2 from the air. The following equations can be used to illustrate charge compensation:
which does not trap any electrons. When a trivalent metallic ion such as Eu3+ is incorporated into a host lattice and substituted for a divalent metallic ion such as Sr2+, then it is difficult to keep a charge balance. In order to maintain the charge balance the host has to capture O2 from the air. The following equations can be used to illustrate charge compensation:
| |
 | (7) |
| |
 | (8) |
| |
 | (9) |
| |
 | (10) |
| |
 | (11) |
In the complex, the [WO4]′ clusters act as electron donors, the vacancy complex  tends to trap electrons or holes and
tends to trap electrons or holes and 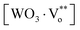 acts as electron traps. PL emission takes place more easily if there are trapped holes or trapped electrons within the band gap. These equations suggest that the oxygen vacancy-trapped electron in the valance band is a necessary requirement for the transition of a valence-band hole in the conduction band.2,18 Therefore, PL of these powders is not due to the structural order–disorder in the lattice. Thus, as previously mentioned, the behavior of this physical property is related to the defects in [WO4] tetrahedron groups.
acts as electron traps. PL emission takes place more easily if there are trapped holes or trapped electrons within the band gap. These equations suggest that the oxygen vacancy-trapped electron in the valance band is a necessary requirement for the transition of a valence-band hole in the conduction band.2,18 Therefore, PL of these powders is not due to the structural order–disorder in the lattice. Thus, as previously mentioned, the behavior of this physical property is related to the defects in [WO4] tetrahedron groups.
CIE study
Fig. 19(a) and (b) show the Commission Internationale de l'Eclairage (CIE) chromaticity diagram for SrWO4:Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) ASP and annealed at 900 °C phosphors excited at 266, 394 and 464 nm, respectively. The calculated CIE color coordinates for all samples are listed in Table 3. The Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) doped SrWO4 show the CIE chromaticity coordinates around the blue, yellow and red regions under different excitations. These results indicate that the color hue can be tuned from blue (point b4) through white (point c3) and finally to red (points a1 and a2) (Fig. 19(a)) in the visible spectral region by changing the doping concentration of Eu3+ or the excitation58 wavelength. It corroborates that under 266 nm excitation, the color coordinates are located in the red region, which is the characteristic region for emissions from Eu3+ and is also in accordance with our observation of the eye-visible luminescence photograph (inset of Fig. 16(a)). The calculated color coordinates of ASP samples vary from (0.29, 0.23) to (0.65, 0.34) providing a large scale tunability of multicolored emission.
 |
| | Fig. 19 The Commission Internationale de l'Eclairage (CIE) chromaticity coordinates for (a) as-prepared and (b) 900 °C annealed samples under different (266, 394 and 464 nm) excitations. | |
Table 4 Parameters obtained after bi-exponential fit to the decay data of as-prepared and 900 °C annealed 2 at.% Eu3+-doped SrWO4 samples under 277 and 395 nm excitations
| Sample |
Excitation (nm) |
I1 (%) |
τ1 (ms) |
I2 (%) |
τ2 (ms) |
τav (ms) |
χ2 |
| ASP |
277 |
62 |
0.38 |
38 |
1.14 |
0.66 |
0.9995 |
| 395 |
54 |
0.29 |
46 |
1.00 |
0.61 |
0.9997 |
| 900 °C |
277 |
57 |
0.76 |
43 |
0.76 |
0.76 |
0.9996 |
| 395 |
49 |
1.25 |
51 |
0.21 |
0.71 |
0.9996 |
On annealing the samples at 900 °C, the value of CIE coordinates increases, thus, the color coordinates also depend on the annealing temperature. In a typical case of 7 at.% Eu3+ doped SrWO4, on annealing the samples, the color perception changes from orange red (presented by point a3, x = 0.58 & y = 0.35) to red (presented by point x3, x = 0.64 & y = 0.33) under 266 nm excitation (Fig. 19(a) and (b)). The annealed sample of SrWO4:Eu3+ has chromaticity coordinates of x = 0.65 and y = 0.34 (λex = 266 nm), which is closer to the standard of NTSC (x = 0.67 & y = 0.33) than the commercial red phosphor Y2O2S:Eu3+ (0.622, 0.351). It suggests that the prepared phosphors can be used in white LEDs.
Decay study
The decay curves for5D0 → 7F2 (613 nm) transition of Eu3+ ions in samples are shown in Fig. 20 and 21 under 277 and 395 nm excitations measured at room temperature. The decay curves for Eu3+ emission can be well fitted by using bi-exponential curve fitting which is expressed as:| | |
I = A1e−t/τ1 + A2e−t/τ2
| (12) |
where, I1 and I2 are the intensities at different time intervals and τ1 and τ2 are their corresponding lifetimes. The bi-exponential fitting of as-prepared and 900 °C annealed 2 at.% Eu3+ doped SrWO4 phosphor under 277 nm excitation are shown in Fig. 20(a) and (b), respectively. Further, the average decay life times can be calculated as:| |
 | (13) |
 |
| | Fig. 20 Bi-exponential fitting to the decay curve (λem = 613 nm) of 2 at.% Eu3+-doped SrWO4 (a) as-prepared and (b) 900 °C annealed samples under 277 nm excitation. | |
 |
| | Fig. 21 Bi-exponential fitting to the decay curve (λem = 613 nm) of 2 at.% Eu3+-doped SrWO4 (a) as-prepared and (b) 900 °C annealed samples under 395 nm excitation. | |
The fitted parameters (I1, τ1, I2, τ2, τav and χ2) obtained using bi-exponential decay equation for ASP and 900 °C annealed 2 at.% Eu3+-doped SrWO4 under 277 and 395 nm are given in Table 4. The average lifetime values for as-prepared and 900 °C annealed samples are found to be 0.666 and 0.765 ms, respectively under 277 nm excitation. These values are in good agreement with the reported values for Eu3+ emission.5,59
In case of 900 °C annealed samples, the lifetime values are higher than that of as-prepared samples. This may be due to reduction in non-radiative rates and removal of O–H bonds from the surface of nano-particles after annealing. The average lifetime upon 277 nm excitation is more than 395 nm excitation, due to the transfer of energy from Eu–O CT band to Eu3+, which is also observed in excitation and emission studies (Fig. 12–15).
Conclusions
Eu3+ (2, 5, 7 and 10 at.%) doped SrWO4 nano-particles were synthesized successfully at low temperature by polyol method under urea hydrolysis. The samples are annealed at 900 °C in order to increase the crystallinity and to remove the dangling bonds present on the surface of the nano-particles. XRD studies corroborate crystalline nature of samples. Raman spectra show the characteristics vibrational modes present in the host lattice. The optical band gap values for ASP and 900 °C annealed 2 at.% Eu3+-doped SrWO4 nano-particles were 4.2 and 3.9 eV, respectively. Luminescence properties were studied under 266, 394 and 464 nm excitations. On annealing the samples, luminescence intensity increases which is related to increase in crystallite size and reduction in non-radiative processes arising from –OH vibrations on the surface of the nano-particles. For ASP and 900 °C annealed SrWO4:Eu3+ (Eu3+ = 2, 5, 7 and 10 at.%) samples, asymmetric values (A21) are in the range of ∼6.0–13.4 corroborating it as a highly red emitter. CIE coordinates are controlled by Eu3+ ion doping and annealing temperature, which dictate the color tunability of the nano-particles. The defect mechanism involved in the system suggested that photoluminescence properties of synthesized nano-powders were not due to the structural order–disorder in the lattice but due to the defects in WO42− tetrahedron groups. Due to the strong red emission in visible region, these nano-particles can be potential candidates to be used in white LEDs.
Acknowledgements
Authors are thankful to UGC DAE Consortium for Scientific Research, Indore for TEM characterization. We are thankful to Sophisticated Instrument Laboratory of the University for providing various characterization facilities. We are also thankful to Prof. S. B. Rai for providing PL facility. One of the authors (Maheshwary) acknowledges the Central Research Fellowship provided by University Grants Commission (UGC), Govt. of India.
References
- K. G. Sharma and N. R. Singh, J. Rare Earths, 2012, 30, 310 CrossRef CAS.
- Z. Ju, R. Wei, X. Gao, W. Liu and C. Pang, Opt. Mater., 2011, 33, 909 CrossRef CAS PubMed.
- P. F. S. Pereira, A. P. de Moura, I. C. Nogueira, M. V. S. Lima, E. Longo, P. C. de Sousa Filho, O. A. Serra, E. J. Nassar and I. L. V. Rosa, J. Alloys Compd., 2012, 526, 11 CrossRef CAS PubMed.
- N. Niu, P. Yang, W. Wang, F. He, S. Gai, D. Wang and J. Lin, Mater. Res. Bull., 2011, 46, 333 CrossRef CAS PubMed.
- A. K. Parchur, R. S. Ningthoujam, S. B. Rai, G. S. Okram, R. A. Singh, M. Tyagi, S. C. Gadkari, R. Tewari and R. K. Vatsa, Dalton Trans., 2011, 40, 7595 RSC.
- P. Yang, C. Li, W. Wang, Z. Quan, S. Gai and J. Lin, J. Solid State Chem., 2009, 182, 2510 CrossRef CAS PubMed.
- Y. Yu-Ling, L. Xue-Ming, F. Wen-Lin, L. Wu-Lin and T. Chuan-Yi, J. Alloys Compd., 2010, 505, 239 CrossRef PubMed.
- I. L. V. Rosa, A. P. A. Marques, M. T. S. Tanaka, F. V. Motta, J. A. Varela, E. R. Leite and E. Longo, J. Fluoresc., 2009, 19, 495 CrossRef CAS PubMed.
- F.-w. Kang, Y.-h. Hu, L. Chen, X. Wang, H. Wu and Z. Mu, J. Lumin., 2013, 135, 113 CrossRef CAS PubMed.
- B. Yan and J. F. Gu, J. Exp. Nanosci., 2009, 4, 301 CrossRef CAS.
- L. Z. Li, B. Yan, L. X. Lin and Y. Zhao, J. Mater. Sci.: Mater. Electron., 2011, 22, 1040 CrossRef CAS.
- Z. J. Zhang, H. H. Chen, X. X. Yang and J. T. Zhao, Mater. Sci. Eng., B, 2007, 145, 34 CrossRef CAS PubMed.
- B. P. Singh, A. K. Parchur, R. S. Ningthoujam, A. A. Ansari, P. Singh and S. B. Rai, Dalton Trans., 2014, 43, 4779 RSC.
- F. Lei and B. Yan, J. Solid State Chem., 2008, 181, 855 CrossRef CAS PubMed.
- Y. Su, G. Li, Y. Xue and L. Li, J. Phys. Chem. C, 2007, 111, 6684 CAS.
- F. Lei, B. Yan and H.-H. Chen, J. Solid State Chem., 2008, 181, 2845 CrossRef CAS PubMed.
- J. C. Sczancoski, L. S. Cavalcante, M. R. Joya, J. W. M. Espinosa, P. S. Pizani, J. A. Varela and E. Longo, J. Colloid Interface Sci., 2009, 330, 227 CrossRef CAS PubMed.
- C. Feldmann, Adv. Funct. Mater., 2003, 13, 101 CrossRef CAS.
- C. Feldmann and H. O. Jungk, Angew. Chem., Int. Ed., 2001, 40, 359 CrossRef CAS.
- D. Kim, S. Jeong and J. Moon, Nanotechnology, 2006, 17, 4019 CrossRef CAS PubMed.
- S. Neeraj, N. Kijima and A. K. Cheetham, Chem. Phys. Lett., 2004, 387, 2 CrossRef CAS PubMed.
- B. P. Singh, A. K. Parchur, R. S. Ningthoujam, A. A. Ansari, P. Singh and S. B. Rai, Dalton Trans., 2014, 43, 4770 RSC.
- S. Phokha, S. Pinitsoontorn, P. Chirawatkul, Y. Poo-arporn and S. Maensiri, Nanoscale Res. Lett., 2012, 7, 425 CrossRef PubMed.
- R. D. Shanon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751 CrossRef.
- A. Kar and A. Patra, Nanoscale, 2012, 4, 3608 RSC.
- J. Rodriguez-Carvajal, FULLPROF a Rietveld and pattern matching analysis program, Laboratoire Leon Brillouin (CEA-CRNS), Paris France Search PubMed.
- M. Yin, A. Willis, F. Redl, N. J. Turro and S. P. O'Brien, J. Mater. Res., 2004, 19, 1208 CrossRef CAS.
- A. K. Parchur, A. I. Prasad, A. A. Ansari, S. B. Rai and R. S. Ningthoujam, Dalton Trans., 2012, 41, 11032 RSC.
- B. P. Singh, A. K. Parchur, R. K. Singh, A. A. Ansari, P. Singh and S. B. Rai, Phys. Chem. Chem. Phys., 2013, 15, 3480 RSC.
- Z. C. Ling, H. R. Xia, D. G. Ran, F. Q. Liu, S. Q. Sun, J. D. Fan, H. J. Zhang, J. Y. Wang and L. L. Yu, Chem. Phys. Lett., 2006, 426, 85 CrossRef CAS PubMed.
- Y. Mao and S. S. Wong, J. Am. Chem. Soc., 2004, 126, 15245 CrossRef CAS PubMed.
- A. P. de Moura, R. C. Lima, M. L. Moreira, D. P. Volanti, J. W. M. Espinosa, M. O. Orlandi, P. S. Pizani, J. A. Varela and E. Longo, Solid State Ionics, 2010, 181, 775 CrossRef CAS PubMed.
- M. R. Nasrabadi, S. M. Pourmortazavi, M. R. Ganjali, S. S. Hajimirsadeghi and M. M. Zahedi, J. Mol. Struct., 2013, 1047, 31 CrossRef PubMed.
- Z. Hou, P. Yang, C. Li, L. Wang, H. Lian, Z. Quan and J. Lin, Chem. Mater., 2008, 20, 6686 CrossRef CAS.
- D. L. Wood and J. Tauc, Phys. Rev. B: Solid State, 1972, 5, 3144 CrossRef.
- Z. Lou and M. Cocivera, Mater. Res. Bull., 2002, 37, 1573 CrossRef CAS.
- M. A. M. A. Maurera, A. G. Souza, L. E. B. Soledade, F. M. Pontes, E. Longo, E. R. Leite and J. A. Varela, Mater. Lett., 2004, 58, 727 CrossRef CAS PubMed.
- K. M. Garadkar, L. A. Ghule, K. B. Sapnar and S. D. Dhole, Mater. Res. Bull., 2013, 48, 1105 CrossRef CAS PubMed.
- A. K. Shahi, B. K. Pandey, R. K. Swarnkar and R. Gopal, Appl. Surf. Sci., 2011, 257, 9846 CrossRef CAS PubMed.
- D. P. Volanti, I. L. V. Rosa, E. C. Paris, C. A. Paskocimas, P. S. Pizani, J. A. Varela and E. Longo, Opt. Mater., 2009, 31, 995 CrossRef CAS PubMed.
-
(a) J. M. Robertson, M. W. v. Tol, W. H. Smits and J. P. H. Heyene, Philips J. Res., 1981, 36, 15 CAS;
(b) H. S. Jang, W. B. Im, D. C. Lee, D. Y. Jeon and S. S. Kim, J. Lumin., 2007, 126, 371 CrossRef CAS PubMed;
(c) M. Shang, C. Li and J. Lin, Chem. Soc. Rev., 2014, 43, 1372 RSC.
- F. B. Cao, Y. W. Tian, Y. J. Chen, L. J. Xiao and Q. Wu, J. Lumin., 2009, 129, 585 CrossRef CAS PubMed.
- J. Wang, X. Jing, C. Yan, J. Lin and F. Liao, J. Lumin., 2006, 121, 57 CrossRef CAS PubMed.
- S. Shi, X. Liu, J. Gao and J. Zhou, Spectrochim. Acta, Part A, 2008, 69, 396 CrossRef PubMed.
- J. Liu, H. Lian and C. Shi, Opt. Mater., 2007, 29, 1591 CrossRef CAS PubMed.
- F. Lei and B. Yan, J. Phys. Chem. C, 2009, 113, 1074 CAS.
- Y. Su, L. Li and G. Li, J. Mater. Chem., 2009, 19, 2316 RSC.
- A. K. Parchur and R. S. Ningthoujam, RSC Adv., 2012, 2, 10859 RSC.
- A. K. Parchur, A. A. Ansari, B. P. Singh, T. N. Hasan, N. A. Syed, S. B. Rai and R. S. Ningthoujam, Integr. Biol., 2014, 6, 53 RSC.
- D. L. Dexter, J. Chem. Phys., 1953, 21, 836 CrossRef CAS PubMed.
- N. Shanta Singh, R. S. Ningthoujam, M. Niraj Luwang, S. Dorendrajit Singh and R. K. Vatsa, Chem. Phys. Lett., 2009, 480, 237 CrossRef PubMed.
- A. K. Parchur, A. I. Prasad, S. B. Rai and R. S. Ningthoujam, Dalton Trans., 2012, 41, 13810 RSC.
- L. R. Singh, R. S. Ningthoujam, V. Sudarsan, I. Srivastava, S. D. Singh, G. K. Dey and S. K. Kulshreshtha, Nanotechnology, 2008, 19, 055201 CrossRef PubMed.
- L. R. Singh, R. S. Ningthoujam, N. Shanta Singh and S. Dorendrajit Singh, Opt. Mater., 2009, 32, 286 CrossRef CAS PubMed.
- G. Blasse, Phys. Lett. A, 1968, 28, 5 CrossRef.
- L. S. Cavalcante, J. C. Sczancoski, J. W. M. Espinosa, J. A. Varela, P. S. Pizani and E. Longo, J. Alloys Compd., 2009, 474, 195 CrossRef CAS PubMed.
- A. P. A. Marques, V. M. Longo, D. de Melo, P. S. Pizani, E. R. Leite, J. A. Varela and E. Longo, J. Solid State Chem., 2008, 181, 1249 CrossRef CAS PubMed.
- L. Wang, Q. Wang, X. Xu, J. Li, L. Gao, W. Kang, J. Shi and J. Wang, J. Mater. Chem. C, 2013, 1, 8033 RSC.
- M. N. Luwang, R. S. Ningthoujam, S. K. Srivastava and R. K. Vatsa, J. Mater. Chem., 2011, 21, 5326 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05903d |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 



















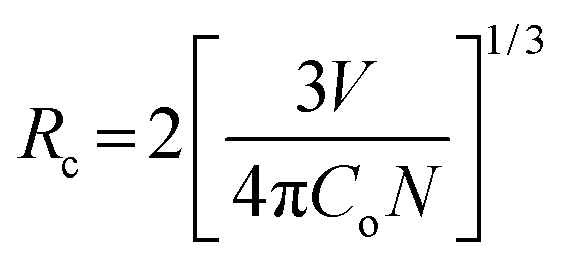
 state, which becomes neutral by capturing electrons,
state, which becomes neutral by capturing electrons,  state, which is singly ionized and the doubly positively charged state in the lattice
state, which is singly ionized and the doubly positively charged state in the lattice  which does not trap any electrons. When a trivalent metallic ion such as Eu3+ is incorporated into a host lattice and substituted for a divalent metallic ion such as Sr2+, then it is difficult to keep a charge balance. In order to maintain the charge balance the host has to capture O2 from the air. The following equations can be used to illustrate charge compensation:
which does not trap any electrons. When a trivalent metallic ion such as Eu3+ is incorporated into a host lattice and substituted for a divalent metallic ion such as Sr2+, then it is difficult to keep a charge balance. In order to maintain the charge balance the host has to capture O2 from the air. The following equations can be used to illustrate charge compensation:




 tends to trap electrons or holes and
tends to trap electrons or holes and  acts as electron traps. PL emission takes place more easily if there are trapped holes or trapped electrons within the band gap. These equations suggest that the oxygen vacancy-trapped electron in the valance band is a necessary requirement for the transition of a valence-band hole in the conduction band.2,18 Therefore, PL of these powders is not due to the structural order–disorder in the lattice. Thus, as previously mentioned, the behavior of this physical property is related to the defects in [WO4] tetrahedron groups.
acts as electron traps. PL emission takes place more easily if there are trapped holes or trapped electrons within the band gap. These equations suggest that the oxygen vacancy-trapped electron in the valance band is a necessary requirement for the transition of a valence-band hole in the conduction band.2,18 Therefore, PL of these powders is not due to the structural order–disorder in the lattice. Thus, as previously mentioned, the behavior of this physical property is related to the defects in [WO4] tetrahedron groups.


