
Preparation and evaluation of hypocrellin A loaded poly(lactic-co-glycolic acid) nanoparticles for photodynamic therapy†
Shan-Shan Qia,
Xi Lina,
Miao-Miao Zhanga,
Shu-Zhen Yana,
Shu-Qin Yu‡
b and
Shuang-Lin Chen*a
aCollege of Life Sciences, Nanjing Normal University, Nanjing 210023, The People's Republic of China. E-mail: chenshuanglin@njnu.edu.cn; Fax: +86 25 8589 1571; Tel: +86 25 8589 1571
bJiangsu Key Laboratory for Supramolecular Medicinal Materials and Applications, College of Life Sciences, Nanjing Normal University, Nanjing 210023, The People's Republic of China. E-mail: yushuqin@njnu.edu.cn; Fax: +86 25 8589 1265; Tel: +86 25 8589 1265
First published on 8th August 2014
Abstract
Hypocrellin A (HA), a perylenequinoid pigment isolated from a traditional Chinese medicinal fungus, exhibits excellent antiviral and antitumor properties. However, its hydrophobicity, photodegeradation and dark cytotoxity hamper its clinical application. The aim of the present study is to prepare and evaluate HA loaded poly(lactic-co-glycolic acid) (PLGA) nanoparticles, which can be dispersed in water. In this present study, an oil-in-water (O/W) emulsion solvent evaporation technique was employed to fabricate hypocrellin A loaded poly(D,L-lactic-co-glycolic) nanoparticles. The physiochemical properties and morphological characteristics were examined. The photostability, uptake and in vitro phototoxicity and dark toxicity toward A549 cells were evaluated. Scanning electron microscopy (SEM) and confocal laser microscopy (CLSM) images combined with dynamic light scattering (DLS) measurements and surface charge (ζ-potential) results showed that the as-prepared HA-loaded nanoparticles had a narrow dispersity with a surface charge of −5.8 mV. The physicochemical properties of the as-prepared nanoparticles were characterized by differential scanning calorimetry (DSC), X-ray powder diffractometry (XRD) and Fourier transform infrared (FTIR) spectroscopy. The encapsulation efficiency and drug content were 55.1% and 5.0%, respectively. UV-vis spectra and the results from photobleaching experiments indicated that encapsulation could enhance the photostability of HA. In vitro experiments demonstrated that HA loaded PLGA nanoparticles were taken up by A549 cells and significantly exhibited reduced dark cytotoxicity, while maintaining excellent anti-tumor property and ROS production ability. These promising results suggested that nanotechnology provide an intriguing possibility for HA being a potent photosensitizer (PS) in clinical photodynamic therapy (PDT).
1. Introduction
Over the past decades, photodynamic therapy (PDT) has been developed rapidly as an alternative treatment strategy for traditional cancer treatment and has gained regulatory approval for the treatment of various diseases such as cancer and macular degeneration.1 This is a two-step progress based on photochemical reactions, including accumulation of photosensitizer (PS) and activation by irradiation.2 After being taken up by the cells in a certain time interval, under proper dose and wavelength of irradiation, the PS can be activated and can generate molecular singlet oxygen (1O2) in the presence of oxygen, which can damage cells via apoptosis or nercrosis.3 Since the PS play such a crucial role in PDT, there are searches for ideal PSs consistently. An ideal PS should have certain properties such as high singlet oxygen quantum yield, excellent solubility in water without forming aggregates and high absorption in the phototherapeutic window.4Hypocrellins, one of the second generation PSs, have attracted increasing attention because of its wide absorption band and excellent light-induced singlet oxygen generating ability.5–7 Hypocrellins are perylenequinoid pigments isolated from the fruiting bodies of the Shiraia bambusicola P. Henn (Fig. 1).8 However, the poor hydrophilicity of hypocrellins highly hampers their clinical applications. Because of their hydrophobicity, hypocrellins tend to aggregate into clusters in the blood and eventually block the vessel.9 Several efforts have been made to overcome this shortcoming, including different types of nanomedical approaches. A variety of studies mainly focused on encapsulating hydrophobic drugs, including hypocrellins, into liposomes to achieve significantly improved dispersion and bioavailability and enhancement of drug efficacy.10–12 However, developmental work on liposomes has been limited due to inherent problems such as low encapsulation efficiency and poor storage stability of a liposome suspension. Many attempts have also been made to deliver hypocrellins via silica nanoparticles, ignoring the deficiencies of silica in biological metabolism.13,14 Moreover, dark toxicity and photodegradation may cause non-specific toxicity in non-targeted cells or organs and counteract the selectivity for PDT.15 Thus, making full use of the characteristics of PLGA to overcome the photodegradation and dark toxicity of hypocrellin and subsequently alleviate unwanted side effects is another intriguing challenge.
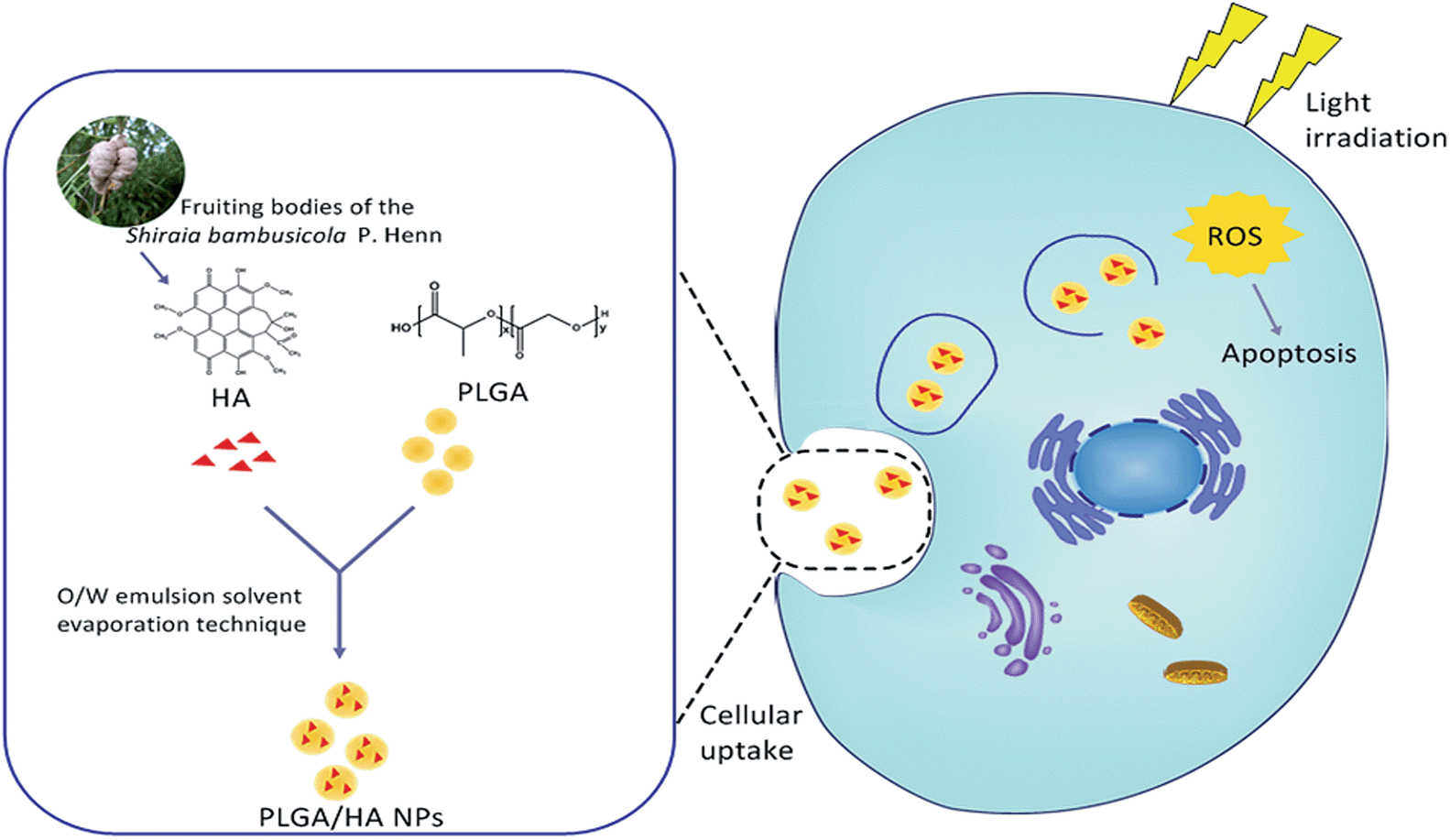 | ||
| Fig. 1 Schematic illustration of the preparation of PLGA/HA NPs and its PDT process. | ||
Recently, various biodegradable delivery systems have been developed for anti-cancer drugs such as PLGA (Fig. 1), hyaluronic acid and saccharide.16–19 Among these biodegradable materials, PLGA has attracted considerable attention. PLGA is one of the commercially available biodegradable materials approved by the Food and Drug Administration (FDA) for human use.20 Emerging evidence has shown that the PLGA nano delivery system also opens new ways for photodynamic therapy because the nanoparticles encapsulated with PSs have the advantages of reduced toxicity, improved dispersibility in plasma, enhanced therapeutic activity and prolonged delivery.21–24 By now, various PSs have been tested in PLGA nano delivery systems for cancer treatment. For instance, 5-aminolevulinic acid (ALA) encapsulated into PLGA gave 65.6 ± 26 nm size particles, an encapsulation efficiency of 65.8% ± 7.2%, and a more effective photo cytotoxicity than free ALA of the same concentration.25 Another study on meso-tetra-(4-hydroxyphenyl) porphyrin (p-THPP) yielded sub-130 nm sized nanoparticles with an increase in the intracellular delivery of p-THPP.26 Hence, PLGA uploading HA could be expected to serve as an intriguing delivery system for PDT. However, few reports about PDT for anticancer in vitro concerning PLGA nanoparticles loaded with hypocrellin A have been reported till now.
In this work, water-dispersible HA loaded PLGA nanoparticles were prepared for the first time applying the oil-in-water (O/W) emulsion solvent evaporation method, and were subsequently characterized by SEM, CLSM, DSC, XRD and FTIR. Furthermore, the photostability, cellular uptake, dark toxicity, phototoxicity and ROS level were also explored.
2. Materials and methods
2.1. Chemicals
Poly(D,L-lactic-co-glycolic acid) (PLGA, L/G = 50/50, MW = 30 kDa) was obtained from Jinan Daigang Biomaterial (Shandong, China). Polyvinyl alcohol (PVA, MW = 31 kDa), 2′,7′-dichlorofluorescin diacetate (DCFH-DA) and dimethylsulfoxide (DMSO) were obtained from Sigma-Aldrich (MO, USA). Dulbecco's minimum essential medium (DMEM) was from Gibico. Cell Counting Kit-8 (CCK-8) was obtained from Dojindo Laboratories (Kumamoto, Japan). Annexin V-FITC Apoptosis Detection kit was obtained from BioVision (USA). Deionized water produced by MilliQ System (Millipore, Paris, France) was utilized throughout the experiments. All other chemicals were of analytical grade.The irradiation power was a light emitting diode (LED) lamp (90 mW cm−2; Cidly Optoelectronic Technology Co., Ltd, China) with a wavelength at 470 nm.
2.2. Isolation of HA
HA was isolated from the fruiting bodies (Fig. 1) of the Shiraia bambusicola P. Henn according to the method reported by Kishi et al. with a slight modification. The air-dried powder of fruit bodies (10.00 g) was extracted with acetone. The organic solvent was evaporated to dryness under vacuum to afford 0.97 g crude extract. After evaporation, the extract was subjected to column chromatography fractionated over silica gel with chloroform–methanol gradient elution. The resulting pigment fraction (260.4 mg) was further separated and purified by preparative thin layer chromatography on silica gel using chloroform–methanol (99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1). Compound 1 (17.3 mg) was subsequently crystallized from acetone. A stock solution of compound 1 at a concentration of 10 mM in DMSO was prepared and stored in the dark at −20 °C.
:1). Compound 1 (17.3 mg) was subsequently crystallized from acetone. A stock solution of compound 1 at a concentration of 10 mM in DMSO was prepared and stored in the dark at −20 °C.
The 1H and 13C NMR data of compound 1 was analyzed by a Bruker Avance III 500 spectrometer (Karlsruhe, Germany). MS spectrum was obtained using an Agilent 1290/6460 HPLC/MS/MS spectrometry system with electrospray ionization (ESI) under positive-ion ionization conditions. FTIR spectra were obtained on a Nexus 670 infrared spectrophotometer (Nicolet, USA) with KBr disks. UV data were recorded on a Hitachi UV-2450 spectrophotometer using methanol as the solvent. Circular dichroism (CD) measurements were performed on a Chirascan™ CD spectrometer (Applied Photophysics, Leatherhead, UK) using methanol as the solvent. XRD data of compound 1 was recorded on a D/max-Rc diffractometer (Ricoh, Japan). DSC measurements were performed on Pyris Diamond calorimeter (Perkin Elmer, USA) at a heating rate of 10 °C min−1.
2.3. Preparation of the PLGA NPs or PLGA/HA NPs
PLGA NPs loaded with HA (PLGA/HA NPs) were prepared by applying an oil-in-water (O/W) emulsion solvent evaporation technique according to our previous study.27 Briefly, 10 mg of HA and 100 mg of PLGA were dissolved in acetone (2.5 mL) for 6 h to form a uniform oil phase. The as-prepared oil phase was then injected into 50 mL of a solution PVA (1%, w/v) at about 11400 rpm under emulsifying conditions in a high-speed homogenizer (IKA, Germany). The residual organic solvent was eliminated by mechanical stirring at a constant speed of 1500 rpm overnight. The NPs were purified by centrifugation (Eppendorf Centrifuge 5418R, Germany) at 13000 rpm for 30 min at 4 °C and washed twice with distilled water to remove the excess drug and emulsifiers. Finally, the freeze-dried powder of PLGA/HA NPs was obtained after lyophilization for 24 h and stored at 4 °C in the dark before use. Blank NPs were prepared without adding HA in the organic phase by employing the same method.
2.4. Characterization of the PLGA/HA NPs
About 1 mg lyophilized powder of nanoparticles was resuspended in deionized water and ultrasonically mixed for 10 minutes to fully disperse the nanoparticles. The particle size and surface charge were measured at room temperature employing a Malvern Zetasizer 3000 laser diffraction grain size analyzer (Worcesterhire, UK).
Thermal analysis was performed using a DSC (Pyris Diamond, Perkin Elmer, USA). Each sample (PLGA, HA, and PLGA/HA NPs) in sealed standard aluminum pans was heated at a rate of 10 °C min−1 from 25 to 270 °C under a dry nitrogen atmosphere.
XRD measurements of PLGA, HA, and PLGA/HA NPs were observed employing a D/max-Rc diffractometer (Ricoh, Japan) with Cu K radiation at a voltage of 40 kV and 200 mA. The scanning rate was 2 min−1 over the interval 3 to 40°.
An FTIR spectrum of each sample (PLGA, HA, and PLGA/HA NPs) was determined by applying the method described above.
000 rpm for 30 min, the absorbance of the supernatant was measured at 464 nm. The amount of the entrapped drug was calculated based on the calibration curve. The measurements were repeated in triplicate, and blank NPs were used as controls.Drug loading content (LC) and drug encapsulation efficiency (EE) were calculated according to the following equations:
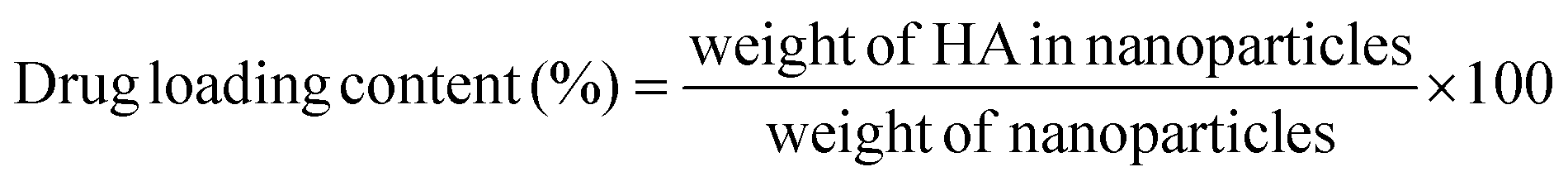 | (1) |
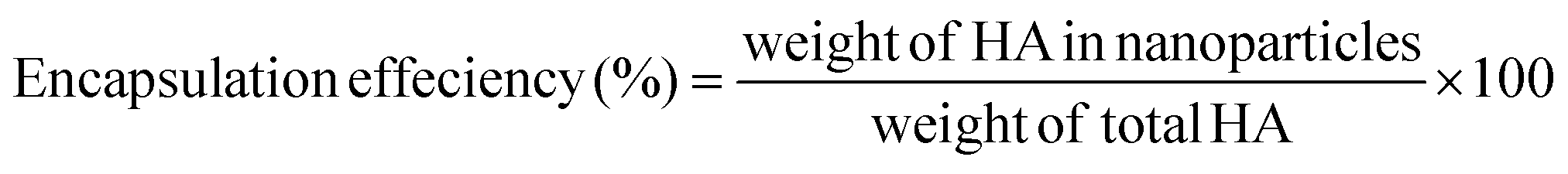 | (2) |
The drug release rate (RR) was calculated according to the following formula:
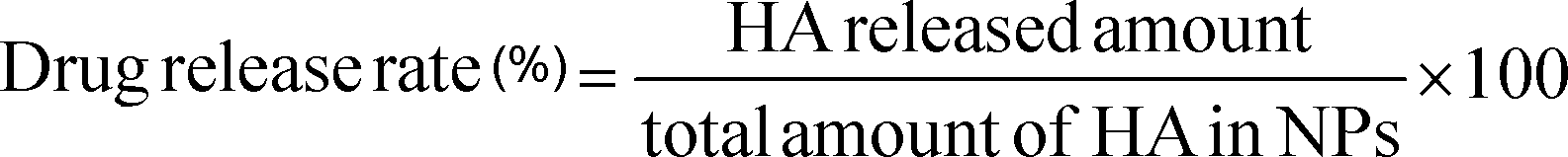 | (3) |
000 rpm for 30 min to obtain the saturated PBS solution of HA. According to Diwu's method, PBS solutions of PLGA/HA NPs was adjusted to give approximate optical density before the photobleaching assay.5 All these samples were illuminated in a standard 1 cm path length quartz cuvette by a high pressure mercury lamp (500 W) and its UV-vis data in the visible region were recorded by a Shimadzu UV-2450 spectrophotometer (Japan).Photobleaching experiments were conducted to compare the photostability of HA before and after encapsulation. After the initial absorbance values were recorded by a UV-vis spectrophotometer, PBS solution of HA or a dispersion of PLGA/HA NPs was exposed to the irradiation of an LED-lamp at a wavelength of 470 nm. At designated time points, 5 mL of each sample was removed and examined at a wavelength of 464 nm. The measurements were repeated in triplicate.
The photostability of HA was calculated as follows:
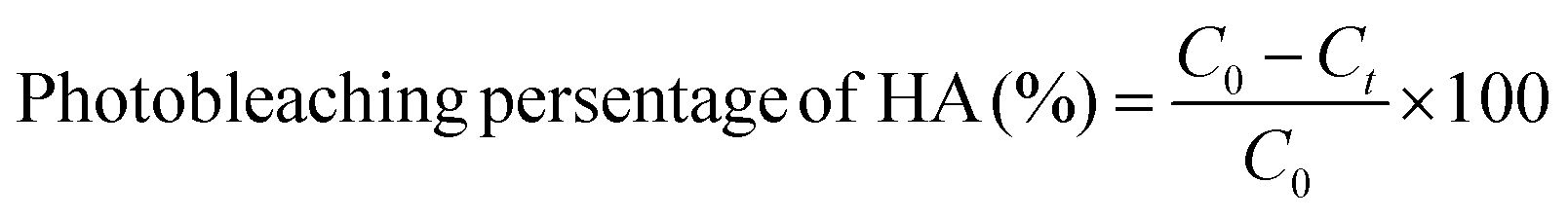 | (4) |
2.5. Cell culture
A549 human lung adenocarcinoma cells line was purchased from American Type Culture Collection (ATCC, USA). A549 cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (v/v) fetal bovine serum (FBS) 100 IU mL−1 penicillin, and 100 μg mL−1 streptomycin at 37 °C in a humidified incubator gassed with 5% CO2.2.6. Photodynamic activity assay and dark cytotoxicity assays
The A549 cells were seeded onto 96-well plates at a density of 2 × 104 cells per cm2 and incubated at 37 °C. After 24 hours, cells were washed with PBS and then incubated at a series of concentrations of HA or PLGA/HA NPs at equivalent HA concentration from 0.5 to 2.5 μM. DMSO (0.1%, v/v) was used as a co-solvent. The cells without any treatment were used as controls. The cells incubated with culture medium containing 0.1% DMSO were used as DMSO controls. After 24 h incubation in the dark, the cells were washed three times with PBS and irradiated by a light emitting diode (LED) lamp with a wavelength at a power peak of 470 nm for 15 min. After irradiation, the cells were incubated in the dark at 37 °C for 18 h before survival assessment. Cell viability was estimated using Cell Counting Kit 8 according to the protocol outlined by the manufacturer. Finally, the absorbance intensity was measured at a wavelength of 450 nm. Following the same protocols, PDT treatments were also performed with cells treated with blank PLGA NPs at doses equivalent to the tested HA loaded NPs to exclude any toxic effect caused by the nano drug carrier alone.The cell viability and cell viability ratio was determined using the following formulae,
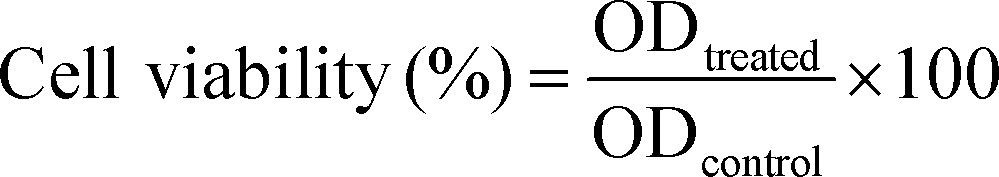 | (5) |
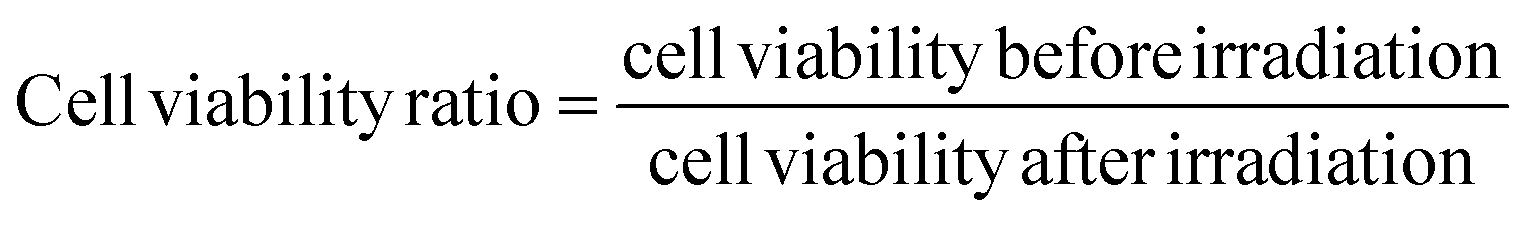 | (6) |
For dark cytotoxicity studies, cells were treated by following exactly the same procedure for cell viability that was mentioned in photodynamic activity assay, except for the light treatment.
2.7. Measurement of reactive oxygen species (ROS)
The generation of intracellular peroxides was determined by applying the probe 2′,7′-dichlorofluorescin diacetate (DCFH-DA). Briefly, approximately 2 × 104 per cm2 cells were seeded in a fluorescence 96-well microplate and incubated overnight for attachment. After washing with PBS, cells were incubated at a series of concentrations of HA or NPs at equivalent HA concentration from 0.5 to 2.5 μM for 4 h. The DMSO control and the empty nanoparticles group, as mentioned above, were also introduced. The cells treated with H2O2 (20 μM) were used as positive controls. For the light group, PDT treatment was conducted as described above. After incubating for 30 min, the cells were washed and incubated with 10 μM of DCFH-DA for 20 min at room temperature without light. Then, the cells were washed twice with PBS to remove the excess probe. Finally, the fluorescence intensity was detected with the excitation and emission wavelengths of 500 and 485 nm, respectively.The ROS level and ROS production ratio was determined using the following formula:
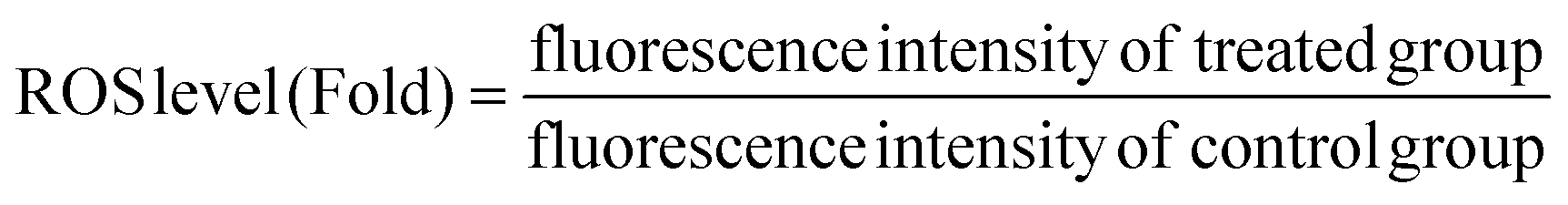 | (7) |
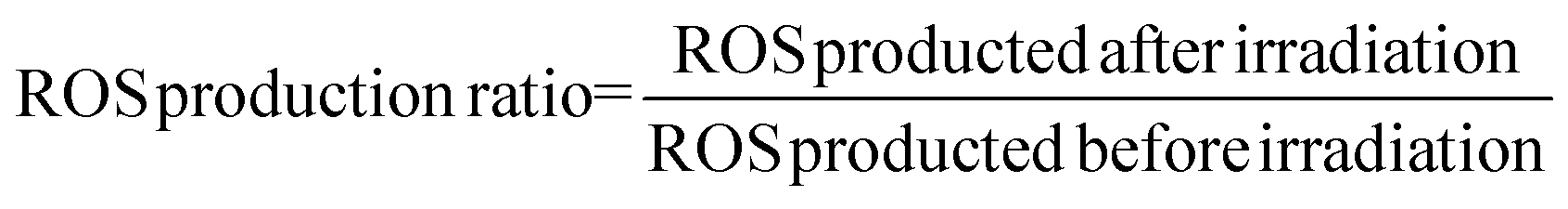 | (8) |
For the dark group, cells were treated by following exactly the same procedure as light group, except for the light treatment.
2.8. Nanoparticle uptake
To determine the cellular uptake of nanoparticles, we seeded 1 × 105 cells per cm2 in 6-well onto the cover glasses. The cells were washed and incubated with PLGA/HA NPs with an equivalent concentration of HA at 2.5 μM for 4 h after attachment. After incubation with nanoparticles, the A549 cells were gently washed three times with PBS. After washing the cells with PBS, the cellular uptake and time-dependent intracellular accumulation of NPs was visualized under a laser scanning confocal microscope (Flowview FV1000, Olympus, Japan).2.9. Measurement of aberrant exposure of phosphatidylserine residues
For the investigation of apoptosis induced by HA or HA loaded NPs, A549 cells were incubated with HA or PLGA/HA NPs (with the equivalent concentration of HA at 2.5 μM) in the dark. After 4 h of incubation, unbound drugs were washed by PBS. For the light group, the cells were exposed to the irradiation of LED-light for 15 min. The dark group was incubated without irradiation. The cells without any treatment were used as controls. After light treatment, the cells of two groups were incubated for 18 h at 37 °C. Cells were harvested and 1 × 106 cells were washed and resuspended with PBS. Eventually, after Annexin V-FITC (fluoresceine isothiocyanate) staining, apoptosis induced by HA or PLGA/HA/NPs were evaluated immediately using flow cytometry. Each sample was analyzed by utilizing a flow cytometry FACS Vantage system (Becton Dickinson, California, USA). Cells that bound Annexin V-FITC were analyzed using 488 nm for excitation and 530 nm for emission.2.10. Statistical analysis
A software package SPSS 17.0 (SPSS Inc., Chicago, USA) was applied for statistical analysis. All the results were expressed as mean ± standard deviation (SD). Student t-test was used for the comparison of the means. P values less than 0.05 were considered to be statistically significant.3. Results and discussion
3.1. Identification of HA
The extraction ratio of compound 1 is 1.73 mg g−1. Compound 1: dark red crystals, mp 216.4 °C (ESI†). UV λmax(MeOH)/nm 267, 341, 464, 540 and 580 nm (Fig. 2A); IR νmax/cm−1 3469 (OH), 1700 (CO) and 1608 (aromatic ring) (Fig. 2B); NMR δH(500 MHz, CDCl3) 15.96 (1H, s, –OH…O–), 15.92 (1H, s, –OH…O–), 6.57 (1H, s, 8-CH = ), 6.55 (1H, s, 5-CH = ), 4.11 (3H × 2, s, –OMe × 2), 4.07(3H × 2, s, –OMe × 2), 3.515 (1H, d, JAB = 12.0 Hz, 13-CHA), 2.635 (1H, d, JAB = 12 Hz, 13-CHB), 3.45 (1H, s, 15-CH), 1.89 (1H, s,18-Me), 1.71 (1H, s, 16-Me) and 1.63 (1H, br. s, 14-OH); δC(500 MHz, CDCl3) 27.0, 30.1, 41.9, 56.5, 56.6, 60.8, 61.7, 62.1, 78.8, 102.0, 102.1, 106.7, 106.9, 117.7, 118.2, 125.0, 127.6, 128.5, 133.2, 134.0, 150.6, 150.9, 167.5, 170.9, 171.8, 179.8, 180.3 and 207.4 (ESI†); ESI-MS m/z 547 ([M + H]+) calcd for C30H26O10 on the basis of MS data (Fig. 2C), 1H and 13C-NMR spectra.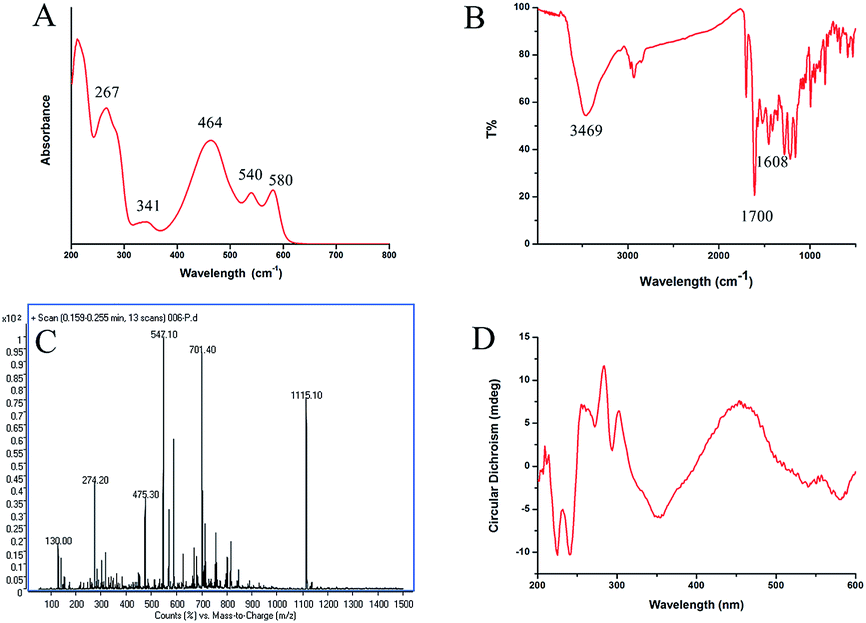 | ||
| Fig. 2 Characterization of HA. (A) UV-vis spectra, (B) FTIR spectra, (C) ESI-MS spectra and (D) CD spectra of HA. | ||
Furthermore, the absolute configuration of compound 1 was also confirmed by comparing CD spectrum with a previous study (Fig. 2D). Compound 1 and isocercosporin (P(S)) had the same axial chirality.
Compound 1 was determined to be hypocrellin A after comparing the 1H-, 13C-NMR data, MS, DSC, FTIR, UV and CD spectrum with those of hypocrellins in literature.8
3.2. Preparation and physicochemical characterization of nanoparticles
Various approaches have been proposed to formulate PLGA NPs by dispersing the preformed polymers, among which emulsion solvent evaporation (O/W) technique was chosen to fabricate polymeric NPs in this work.28 Polyvinyl alcohol (PVA) was applied as the emulsifying agent during the process because the nanoparticulate carrier formulated by employing this emulsifier are comparatively homogeneous, small-sized and redispersible in aqueous solution.29,30SEM images of the HA loaded NPs is shown in Fig. 3, suggesting that the NPs had a distribution over the range of about 20–200 nm with a spherical shape. Inset panel of Fig. 3 was in consistent with the particle size in the SEM. The ζ potential of HA loaded NPs and blank NPs was −5.8 and −7.3 mV, respectively, which might be attributed to the dissociation of the carboxylic end groups of PLGA (Table 1).
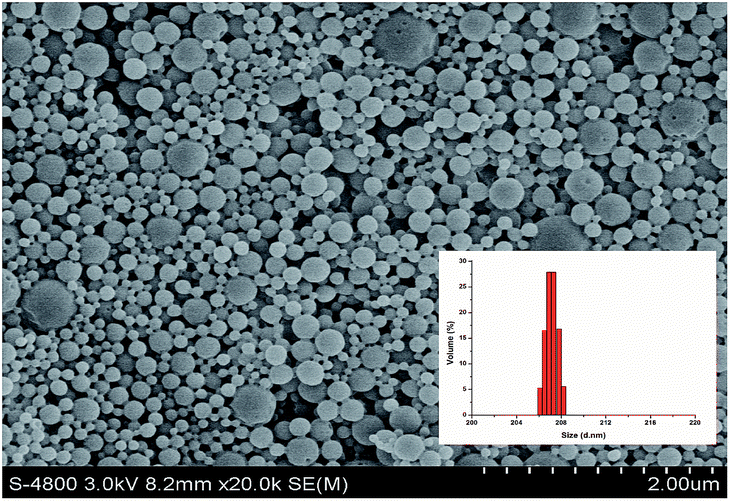 | ||
| Fig. 3 Morphology property and size distribution of PLGA/HA NPs. | ||
| EE (%) | LC (wt%) | ζ (mV) | |
|---|---|---|---|
| Blank NPs | — | — | −7.3 |
| PLGA/HA NPs | 55.1 ± 1.05 | 5.0 ± 0.10 | −5.8 |
In previous studies, the surface charge and size of the nanoparticles played a critical role concerning cellular uptake.31,32 A potential advantage of nano-scale particle size is that it could contribute to increased residence time in blood and the improve biodistribution of the carrier via the EPR effect, which subsequently brings a passive selective tumor targeting and delivery.33 In addition, as cationic particles that can bind more efficiently to most cell surfaces before reaching the target location, it might be crucial to maintain the nano-size carriers either anionic or neutral for less untargeted binding and thereby reduce unexpected side effects.32
DSC was employed to find out the crystal transformation of the nanoparticles. As demonstrated in Fig. 4A, the melting peak of crystalline HA appeared at 216.4 °C, which is consistent with published literature.8 PLGA polymer gives rise to a small peak around 45 °C, corresponding to its glass transition temperature. However, no notable peak was found from the curves of the HA loaded NPs except the same glass transition temperature at around 45 °C, indicating that HA converted from crystalline to amorphous from.
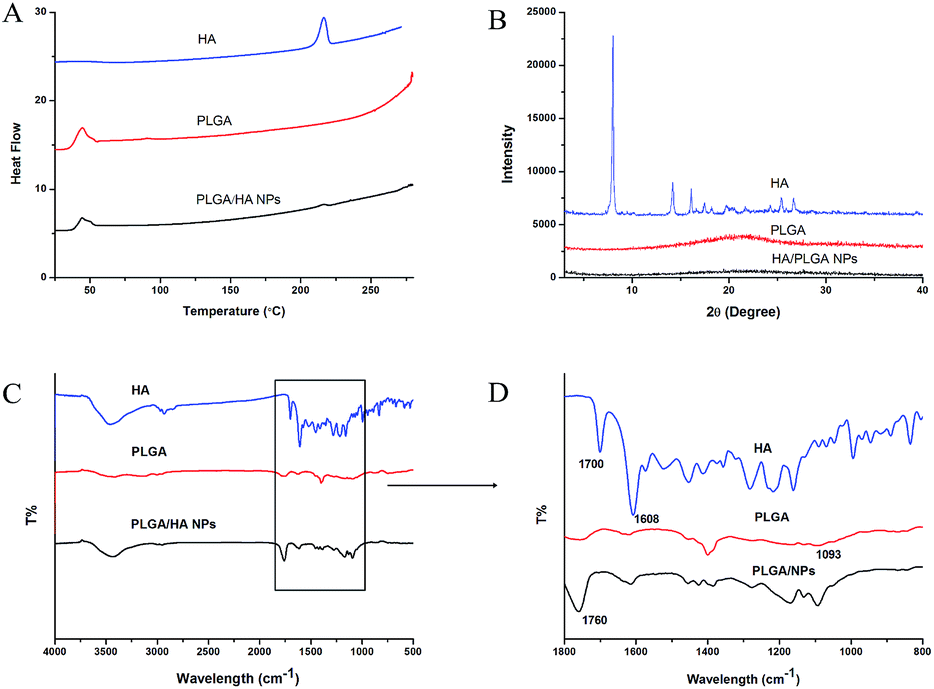 | ||
| Fig. 4 Physicochemical characterization of HA, PLGA and PLGA/HA NPs. (A) DSC curves, (B) XRD patterns, and (C) and (D) FTIR spectra of HA, PLGA and PLGA/HA NPs. | ||
X-ray diffraction (XRD) measurements were carried out to observe the interactions between HA and PLGA. Fig. 4B clearly shows that the characteristic peak of HA appeared at 8.020°,14.180°, 16.100°, 25.380° and 26.680°, indicating the crystalline structure of HA. However, these characteristic peaks disappeared in the curve of NPs, which suggested that HA no longer existed in a crystalline form and was successfully entrapped into the NPs.
To learn more about the physicochemical characteristics of PLGA/HA NPs, FTIR was carried out. The FTIR spectra of HA, PLGA and PLGA/HA NPs is shown in Fig. 4C and D. The characteristic band of PLGA at 1685 cm−1 and 3413 cm−1 corresponds to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group and O–H group, respectively. For the spectra of HA, the sharp peak at 1608 cm−1 is attributed to the characteristic band of the aromatic ring of HA. The characteristic peak of HA at 1700 cm−1 is attributed to the CO stretching vibration, which was slightly shifted to 1760 cm−1 in the spectrum of the NPs. This could be attributed to the formation of intermolecular hydrogen bonds between the CO group of PLGA and the O–H group of HA.
O group and O–H group, respectively. For the spectra of HA, the sharp peak at 1608 cm−1 is attributed to the characteristic band of the aromatic ring of HA. The characteristic peak of HA at 1700 cm−1 is attributed to the CO stretching vibration, which was slightly shifted to 1760 cm−1 in the spectrum of the NPs. This could be attributed to the formation of intermolecular hydrogen bonds between the CO group of PLGA and the O–H group of HA.
From the results presented above, we can conclude that PLGA/HA/NPs were successfully formulated and negatively charged with a spherical shape and a narrow size distribution. Meanwhile, the SEM and CLSM study proved that the PLGA/HA NPs were nano-scale, which could improve water dispersibility and biocompatibility.
3.3. Drug loading content and encapsulation efficiency
To evaluate the drug encapsulation efficiency (EE) and drug loading content (LC) of PLGA/HA NPs, the UV-vis spectrophotometer method was used. The absorbance values at 464 nm were found to be linearly correlated with HA concentrations ranging from 0 to 24 μg mL−1 with a correlation coefficient of 0.999. As shown in Table 1, the drug loading content and encapsulation efficiency of the PLGA/HA NPs were 5.0% and 55.1%, respectively.3.4. In vitro release of HA from PLGA/HA NPs
The study of the in vitro release of HA from HA loaded NPs over a seven-day period is presented in Fig. 5 pH 7.4 and 6.5 PBS were selected as the release phase, simulating both physiological and tumor extracellular environments.34 As depicted in Fig. 5, in both the cases, the drug release kinetics displayed a biphasic release with fast release in the first 24 hours followed by a slower release for the rest of the experiment. In the first 24 hours, 28.2% and 27.4% of HA were released at pH 7.4 and 6.5, respectively. By the end of the test time, a maximum of 49.6% of HA released from NPs was achieved at pH 6.5, which were considerably faster than that at pH 7.4 (38%).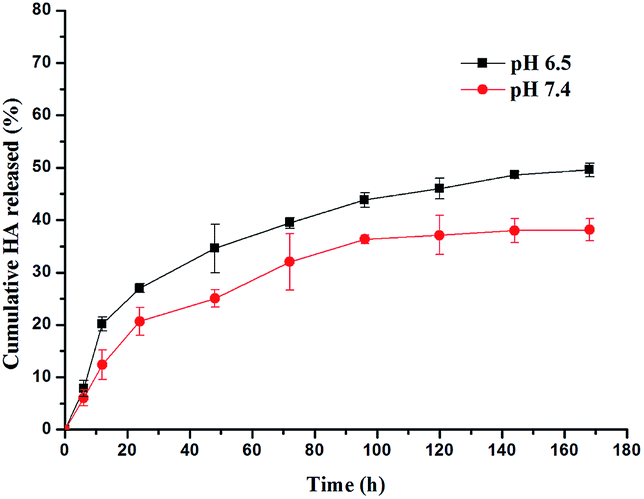 | ||
| Fig. 5 In vitro cumulative release of HA from the PLGA/HA NPs. | ||
The fast release rate of HA during the first stage could be attributed to the release of HA from the external surface of the PLGA/HA NPs. The entrapped HA in the PLGA substrate of nanocarriers is then released at a significantly slower rate, which is mainly due to diffusion from the polymeric substrate, thus giving a nano-formulation of sustained release with remarkable photodynamic effects.35 Another noticeable phenomenon is that the release rate of HA from the nanoparticles at pH 6.5 was higher than that in an artificial blood physiological environment (pH 7.4), indicating that the PLGA/HA NPs were stable with a less non-specific release while circulating in the blood. This characteristic may encourage HA to release more in a tumor area via passive targeting. It is widely believed that tumor tissue has some unique anatomical and pathophysiological vasculature characteristics, such as high vascular density and an ineffective lymphatic system, which cause an “enhanced permeability and retention” (EPR) effect. Maeda and his group first managed to design NPs to achieve tumor targeting delivery by taking advantage of the EPR effect.36 The enhanced accumulation of NPs, combined with its release kinetic and good water-dispersibility, could contribute to a favorable biodistribution of HA and more selective targeting of HA loaded nanoparticles to tumors.
3.5. Photophysical characterization and photostability study
Freeze-dried HA loaded PLGA NPs was found to be dispersible in water, while free HA can hardly dissolve in water matrices without the help of other solvents (Fig. 6A). To investigate the absorption spectrum of HA and PLGA/HA NPs, a UV-vis spectrophotometer was applied. As shown in the inset panel of Fig. 6B, free HA in PBS (pH 7.4) has three characteristic absorbance bands at 465 nm, 534 nm and 586 nm. After encapsulation into PLGA, the characteristic absorbance band at 464 nm was slightly red-shifted to 473 nm. The UV spectra suggested that HA preserved its photochemical characteristics after nanoencapsulation.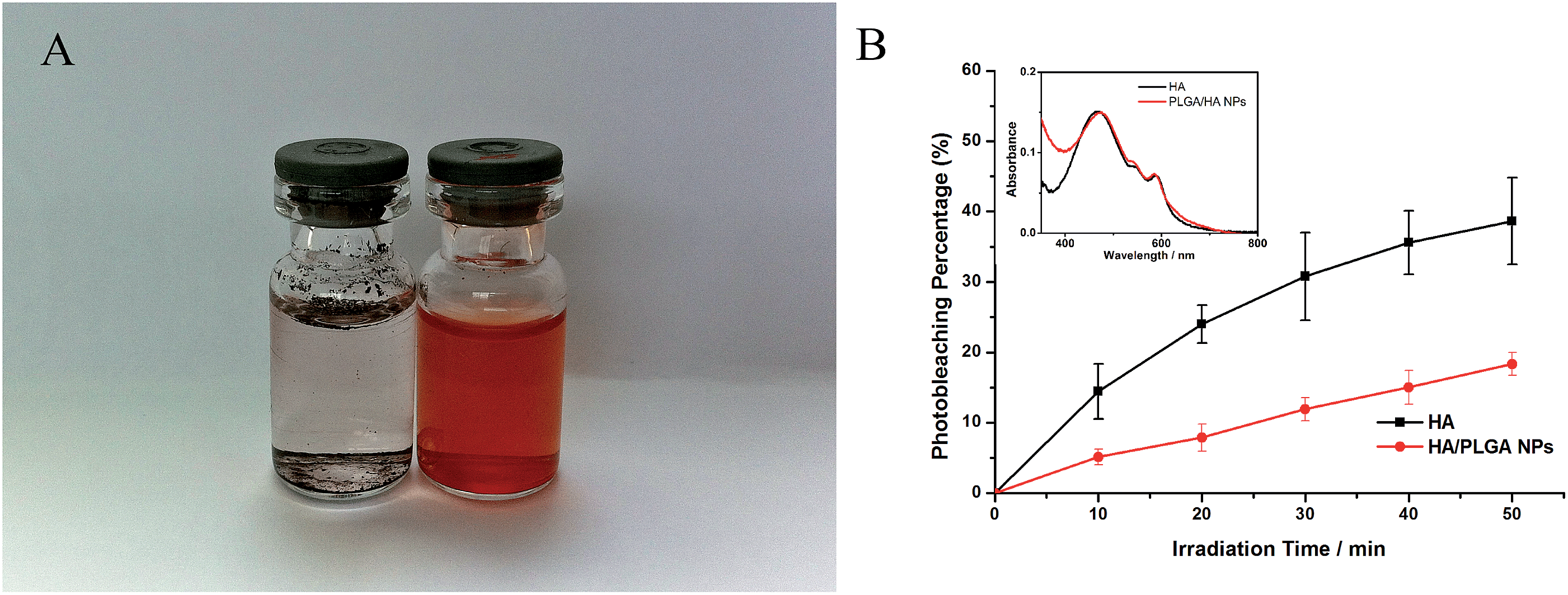 | ||
| Fig. 6 Dispersibility, photophysical characterization and photostability study. (A) Photo images of HA and PLGA/HA NPs in water. (B) Photobleaching experiments of HA and PLGA/HA NPs (inset picture: absorption spectra of HA and PLGA/HA NPs in pH 7.4 buffer). | ||
To observe the photostability of HA and PLGA/HA NPs, a photobleaching experiment was introduced. After the PBS dispersion of HA loaded PLGA NPs or solution of HA were irradiated by a LED-lamp with a wavelength at 470 nm, the intensity of the absorption peaks decreased with the irradiation time (Fig. 6B). At the last time point, photobleaching efficiencies of HA loaded NPs and free HA are 18.34% and 38.64%, respectively, indicating that HA encapsulated into PLGA had improved photostability than free HA because of the protection from the PLGA.
Most PSs, including HA, are not photostable. In simple media, along with complex dispersion or solution, photo-induced bleaching of PSs reduces their fluorescence intensity and initial absorption, which consequently lowers the photosensitive effect. According to our study, the photobleaching experiments of HA and PLGA/HA NPs dispersions indicated that the absorption intensity of HA decreased notably in water, which is in consistent with Zhou's study.13 For this phenomenon, the probable reason is either non-radiative decay promoted by drug-solvent interaction or concentration quenching processes that come from self-aggregation of HA. After encapsulated into PLGA, the inside HA molecules (non-polar drug) can be protected against the exposure to polar media such as aqueous solutions. The protection effect provided by PLGA nanoparticles offers enhanced photostability of HA against photobleaching.
3.6. Cell viability and ROS detection
The in vitro dark cytotoxicity and photodynamic activity of free HA and PLGA/HA NPs is shown in Fig. 7A and B, respectively. Without irradiation, a sharp decrease in cell viability was observed with the HA concentration from 0.5 μM to 2.5 μM (Fig. 7A). The PLGA/HA NPs were nearly non-toxic at all the tested concentrations when incubated with cells in the dark, whereas at concentrations above 2 μM, the cytotoxicity delivered by free HA was significantly higher.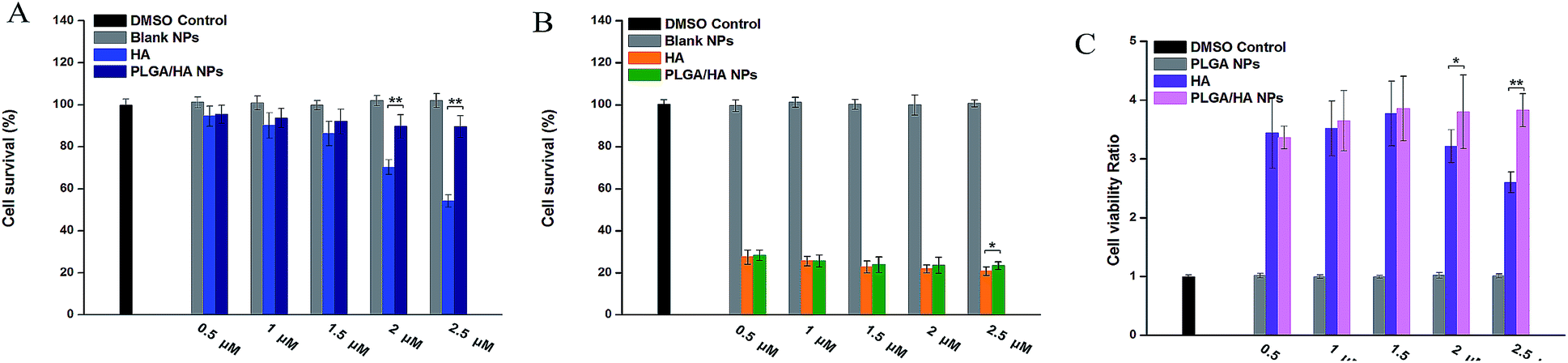 | ||
| Fig. 7 Dark cytotoxicity assays (A) and photodynamic activity assays (B) on A549 cells. *p < 0.05; **p < 0.01 (t-test). | ||
Unlike dark cytotoxicity, when LED light was applied, the situation became different. Both the free HA and NPs were highly toxic to A549 cells when exposed to the LED light with a slight tendency to decrease this property as the concentrations were increased. Even at the minimum concentration of 0.5 μM the cell viability was still 20.8% and 23.4% (Fig. 7B).
Since the first time DCFH-DA was applied in the hydrogen peroxide detection assay, it became popular to use it as a indicator for evaluating the formation of intracellular reactive oxygen species.37 The nonpolar probe DCFH-DA crosses cell membranes and is enzymatically hydrolyzed to the nonfluorescent dichlorofluorescin (DCFH). In the presence of reactive species, nonfluorescent DCFH would be oxidized to dichlorofluorescein (DCF), which emits a strong fluorescence. The ROS detection data is shown in Fig. 8. Without irradiation, the level of ROS in the HA treated group was significantly higher than PLGA/HA NPs treated group at a concentration of 2.5 μM. After exposure to LED light, the HA treated group at five tested concentrations produced slightly more ROS than PLGA/HA NPs, which is basically consistent with the cell toxicity results.
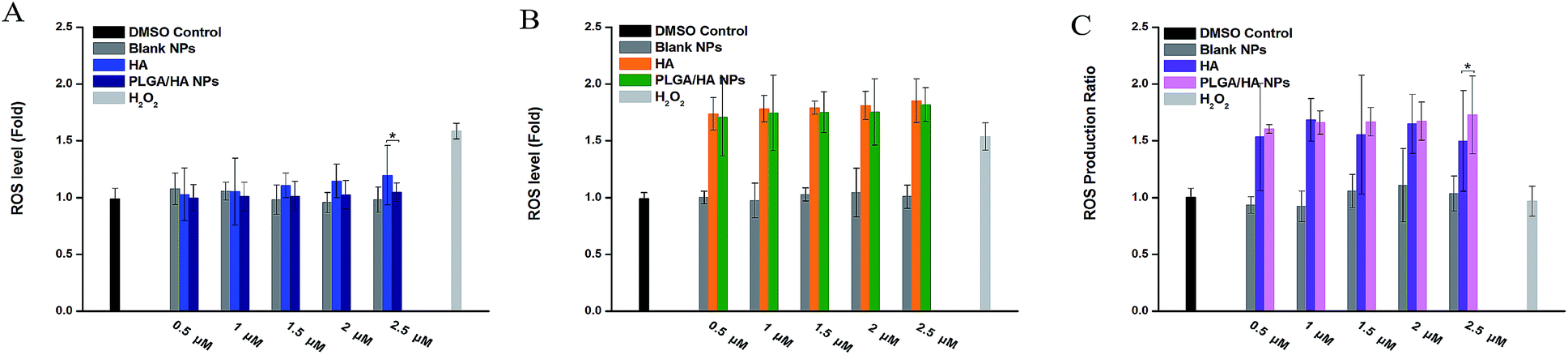 | ||
| Fig. 8 ROS level before PDT (A), after PDT (B) and ROS production ratio (C) on A549 cells. *p < 0.05; **p < 0.01 (t-test). | ||
Both data or the results of cell viability ratio (Fig. 7C) and ROS production ratio (Fig. 8C) suggest that the nano-formulation of HA had better toxicity selectivity toward A549 cells at a relatively high concentration.
3.7. Cellular uptake assays
The cellular uptake of HA or HA loaded PLGA NPs into A549 cells was visualized using a laser scanning confocal microscope (Fig. 9). The fluorescence of HA excited by the 543 nm laser makes it readily detectable by CLSM after cellular uptake. The CLSM images showed that PLGA/HA NPs emitting red fluorescence was mainly accumulated in the cytoplasm. Furthermore, the nano-formulated HA was internalized more actively than free HA, presumably through an endocytic process according to former studies conducted by Panyam et al.38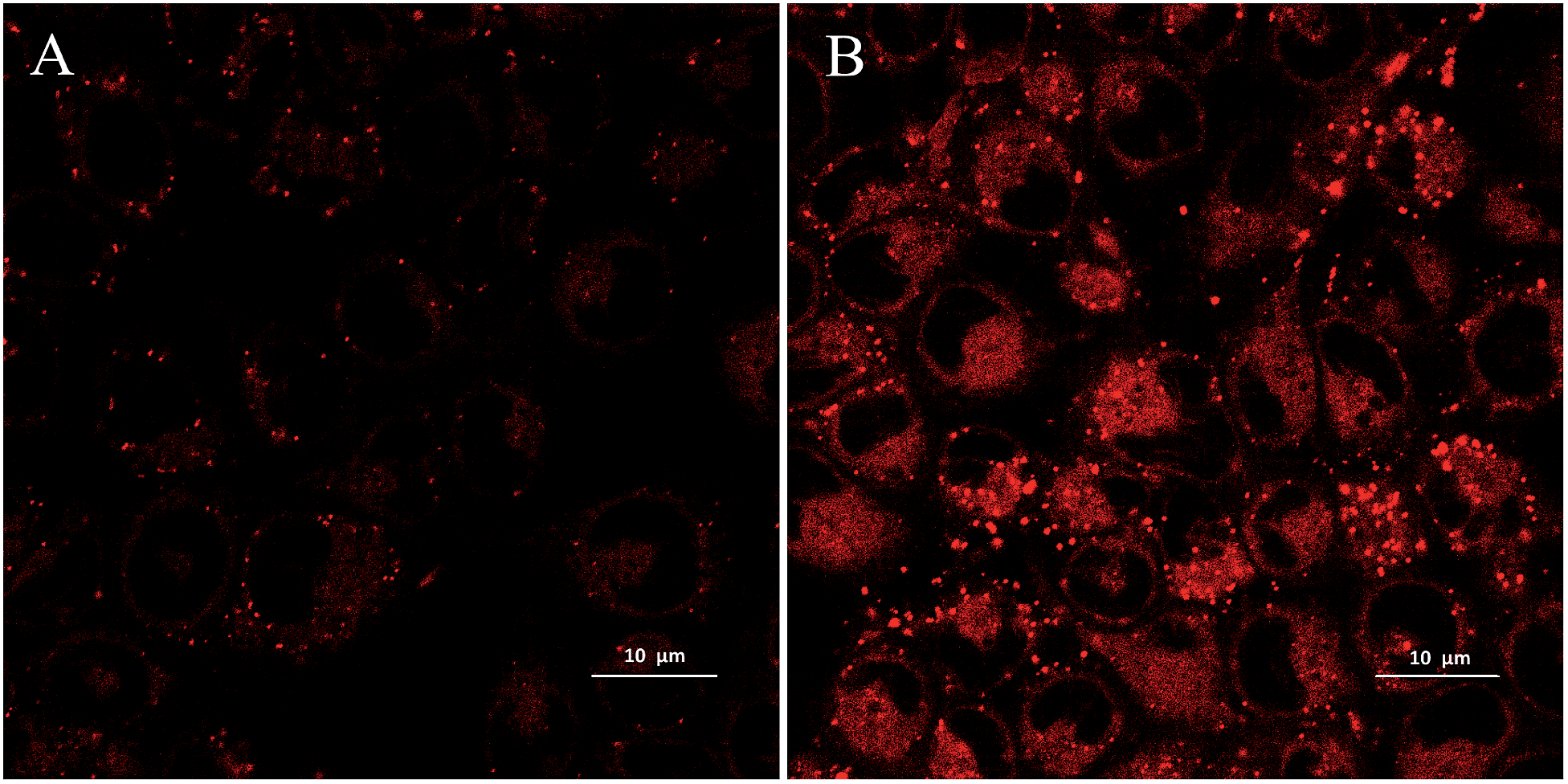 | ||
| Fig. 9 Confocal laser microscopy images of A549 cells incubated with HA (A) and PLGA/HA/NPs (B) with equivalent HA concentration (2.5 μM) (magnification 100×). | ||
Fei et al. proved that in the absence of illumination, HA could still directly result in the death of HeLa cells, indicating that there are potential dark toxicity risks.39 Once the NPs were internalized by endocytosis, PLGA/HA NPs were in the endosomal compartments and released HA from PLGA in a sustained manner. The release kinetics, combined with the cellular uptake mechanism of PLGA/HA NPs may help to alleviate the negative effect of hypocrellin A in the non-targeted area, and lead to a lower dark toxicity. When light was applied, the activated HA released from nanoparticles might eventually kill A549 cells. However, further evaluation is required to find out the possible cell death mechanism.
3.8. Analysis of apoptosis
Since Dougherty and his colleague first demonstrated that PDT can be harnessed to kill tumor cells in 1978, various PSs have been investigated in vitro or in vivo to understand the mode of cell death.40 According to the latest studies, the mechanism of cell death by PDT may be either apoptosis or necrosis, which largely depends on the photochemistry and photophysical property of the PSs.2,41 Recently, an increasing number of studies have suggested that hypocrellin is a strong inducer of cell apoptosis. Moreover, reactive oxygen species (ROS) generated by PS in the presence of irradiation and oxygen might play a pivotal role in this apoptosis process, especially singlet oxygen.42 From the above-mentioned results it can be seen that PLGA/HA NPs might induce cell death via a similar mechanism.To further confirm our speculation, an apoptosis assay was carried out. Phosphatidylserine externalization is one of the critical events related to apoptotic death in tumor cells in response to various kinds of stimuli such as PS. Phosphatidylserine exposure on the exterior surface of A549 cells was evaluated using flow cytometry with the FITC-labeled annexin V staining following the manufacturer's instructions. The concentration of 2.5 μM was chosen because of its better toxicity selectivity. After activated by LED-light, both HA and PLGA/HA NPs increased the expression of phosphatidylserine on the interior surface of cell membrane (49.38% and 46.73%), indicating loss of Phospholipid asymmetry (Fig. 10). However, without irradiation, the phosphatidylserine externalization of cells incubated with HA was evidently more than the cells incubated with PLGA/HA NPs with an equivalent concentration of HA, confirming a significantly stronger cellular destruction of free HA.
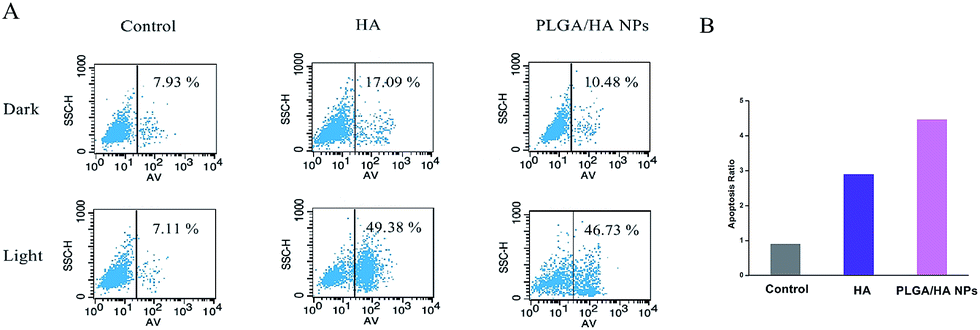 | ||
| Fig. 10 Phosphatidylserine externalization assays on A549 cells (A) and the apoptosis ratio with or without irradiation (B). | ||
4. Conclusions
In summary, novel water-dispersible HA loaded PLGA NPs have been fabricated by applying oil-in-water (O/W) emulsion solvent evaporation technique based on the superior characteristics of PLGA, which is biodegradable and biocompatible. The HA loaded PLGA nanoparticles were narrowly distributed with a spherical shape and a negative surface charge. The resulting HA-loaded PLGA nanoparticles have enhanced photostability and can be actively internalized by A549 cells. A notable decrease in dark cytotoxicity was observed, which is likely due to negative surface charge and the drug release of nanoparticles occurred at a preferred lower pH. Based on the above-mentioned results, it can be concluded that the PLGA nanodrug delivery system could provide a promising future for HA in the field of anticancer PDT.Acknowledgements
This study was supported by National Key Technology Research and Development Program of the Ministry of Science and Technology of China (Project no. 2012BAD36B0502).Notes and references
- M. Triesscheijn, P. Baas, J. H. M. Schellens and F. A. Stewart, Oncologist, 2006, 11, 1034–1044 CrossRef CAS PubMed.
- C. A. Robertson, D. H. Evans and H. Abrahamse, J. Photochem. Photobiol., B, 2009, 96, 1–8 CrossRef CAS PubMed.
- N. L. Oleinick, R. L. Morris and I. Belichenko, Photochem. Photobiol. Sci., 2002, 1, 1–21 CAS.
- E. Paszko, C. Ehrhardt, M. O. Senge, D. P. Kelleher and J. V. Reynolds, Photodiagn. Photodyn. Ther., 2011, 8, 14–29 CrossRef CAS PubMed.
- Z. Diwu, C. Zhang and J. W. Lown, Anti-Cancer Drug Des., 1993, 8, 129–143 CAS.
- Z. Diwu, Photochem. Photobiol., 1995, 61, 529–539 CrossRef CAS PubMed.
- Z. Zhiyi, W. Nenghui, W. Qian and L. Meifan, Free Radical Biol. Med., 1993, 14, 1–9 CrossRef CAS.
- T. Kishi, S. Tahara, N. Taniguchi, M. Tsuda, C. Tanaka and S. Takahashi, Planta Med., 1991, 57, 376–379 CrossRef CAS PubMed.
- F. Wang, L. Zhou, J. H. Zhou, X. T. Gu and Y. Y. Feng, J. Therm. Anal. Calorim., 2010, 102, 69–74 CrossRef CAS.
- D. Bai, X. Xia, C. M. Yow, E. S. Chu and C. Xu, Eur. J. Pharmacol., 2011, 650, 496–500 CrossRef CAS PubMed.
- C. Yu, S. Chen, M. Zhang and T. Shen, Photochem. Photobiol., 2001, 73, 482–488 CrossRef CAS.
- Y. Liu, J. Fang, Y. J. Kim, M. K. Wong and P. Wang, Mol. Pharm., 2014, 11, 1651–1661 CrossRef CAS PubMed.
- L. Zhou, Y. W. Ning, S. H. Wei, Y. Y. Feng, J. H. Zhou, B. Y. Yu and J. Shen, J. Mater. Sci.: Mater. Med., 2010, 21, 2095–2101 CrossRef CAS PubMed.
- Z. B. Li, J. G. Wang, J. R. Chen, W. H. Lei, X. S. Wang and B. W. Zhang, Sci. China: Chem., 2010, 53, 1994–1999 CrossRef CAS PubMed.
- A. Master, M. Livingston and A. Sen Gupta, J. Controlled Release, 2013, 168, 88–102 CrossRef CAS PubMed.
- B. Sivakumar, R. G. Aswathy, Y. Nagaoka, S. Iwai, K. Venugopal, K. Kato, Y. Yoshida, T. Maekawa and D. N. Sakthi Kumar, RSC Adv., 2013, 3, 20579 RSC.
- S. K. Tripathi, S. Gupta, K. C. Gupta and P. Kumar, RSC Adv., 2013, 3, 15687 RSC.
- Z. Yu, R. M. Schmaltz, T. C. Bozeman, R. Paul, M. J. Rishel, K. S. Tsosie and S. M. Hecht, J. Am. Chem. Soc., 2013, 135, 2883–2886 CrossRef CAS PubMed.
- C. Bhattacharya, Z. Yu, M. J. Rishel and S. M. Hecht, Biochemistry, 2014, 53, 3264–3266 CrossRef CAS PubMed.
- F. Danhier, E. Ansorena, J. M. Silva, R. Coco, A. Le Breton and V. Préat, J. Controlled Release, 2012, 161, 505–522 CrossRef CAS PubMed.
- M. Zeisser-Labouèbe, N. Lange, R. Gurny and F. Delie, Int. J. Pharm., 2006, 326, 174–181 CrossRef PubMed.
- A. J. Gomes, A. S. Faustino, A. E. Machado, M. E. Zaniquelli, T. de Paula Rigoletto, C. N. Lunardi and L. O. Lunardi, Drug Delivery, 2006, 13, 447–454 CrossRef CAS PubMed.
- C. L. da Silva, J. O. Del Ciampo, F. C. Rossetti, M. V. Bentley and M. B. Pierre, Photochem. Photobiol., 2013, 89, 1176–1184 CrossRef CAS PubMed.
- A. Vargas, N. Lange, T. Arvinte, R. Cerny, R. Gurny and F. Delie, J. Drug Targeting, 2009, 17, 599–609 CrossRef CAS PubMed.
- L. Shi, X. Wang, F. Zhao, H. Luan, Q. Tu, Z. Huang, H. Wang and H. Wang, Int. J. Nanomed., 2013, 2669 CrossRef PubMed.
- Y. N. Konan, J. Chevallier, R. Gurny and E. Allemann, Photochem. Photobiol., 2003, 77, 638–644 CrossRef CAS.
- X. Xie, Q. Tao, Y. Zou, F. Zhang, M. Guo, Y. Wang, H. Wang, Q. Zhou and S. Yu, J. Agric. Food Chem., 2011, 59, 9280–9289 CrossRef CAS PubMed.
- C. E. Astete and C. M. Sabliov, J. Biomater. Sci., Polym. Ed., 2006, 17, 247–289 CrossRef CAS PubMed.
- S. K. Sahoo, J. Panyam, S. Prabha and V. Labhasetwar, J. Controlled Release, 2002, 82, 105–114 CrossRef CAS.
- C. Wischke, Y. Zhang, S. Mittal and S. P. Schwendeman, Pharm. Res., 2010, 27, 2063–2074 CrossRef CAS PubMed.
- C. He, Y. Hu, L. Yin, C. Tang and C. Yin, Biomaterials, 2010, 31, 3657–3666 CrossRef CAS PubMed.
- K. Y. Win and S. S. Feng, Biomaterials, 2005, 26, 2713–2722 CrossRef CAS PubMed.
- S. Acharya and S. K. Sahoo, Adv. Drug Delivery Rev., 2011, 63, 170–183 CrossRef CAS PubMed.
- J. Wang, J. Sun, Q. Chen, Y. Gao, L. Li, H. Li, D. Leng, Y. Wang, Y. Sun, Y. Jing, S. Wang and Z. He, Biomaterials, 2012, 33, 6877–6888 CrossRef CAS PubMed.
- T. Niwa, H. Takeuchi, T. Hino, N. Kunou and Y. Kawashima, J. Controlled Release, 1993, 25, 89–98 CrossRef CAS.
- M. S. Cartiera, K. M. Johnson, V. Rajendran, M. J. Caplan and W. M. Saltzman, Biomaterials, 2009, 30, 2790–2798 CrossRef CAS PubMed.
- A. S. Keston and R. Brandt, Anal. Biochem., 1965, 11, 1–5 CrossRef CAS.
- J. Panyam, W. Z. Zhou, S. Prabha, S. K. Sahoo and V. Labhasetwar, FASEB J., 2002, 16, 1217–1226 CrossRef CAS PubMed.
- X. F. Fei, J. Chen, K. Y. Zheng, W. Wei, S. J. Sun, L. Wang, L. Ma, C. Li and L. R. Teng, Chem. Res. Chin. Univ., 2006, 22, 772–775 CrossRef CAS.
- T. J. Dougherty, J. E. Kaufman, A. Goldfarb, K. R. Weishaupt, D. Boyle and A. Mittleman, Cancer Res., 1978, 38, 2628–2635 CAS.
- D. Lihuan, Z. Jingcun, J. Ning, W. Guozeng, C. Yiwei, L. Wei, Q. Jing, Z. Yuanfang and C. Gang, Lasers Surg. Med., 2014, 46, 319–334 CrossRef PubMed.
- Y.-H. Kim, Oncol. Rep., 2013, 30, 856–862 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05796a |
| ‡ This author is the second corresponding author. |
| This journal is © The Royal Society of Chemistry 2014 |