
Scalable production of transition metal disulphide/graphite nanoflake composites for high-performance lithium storage†
Zhi-Qiang Duan‡
a,
Yan-Chun Sun‡b,
Yi-Tao Liu*a,
Xu-Ming Xie*a and
Xiao-Dong Zhu*c
aLaboratory of Advanced Materials (Ministry of Education), Department of Chemical Engineering, Tsinghua University, Beijing 100084, China. E-mail: liu-yt03@mails.tsinghua.edu.cn; xxm-dce@mail.tsinghua.edu.cn; Fax: +86 10 62784550; Tel: +86 10 62773607
bHeilongjiang River Fishery Research Institute, Chinese Academy of Fishery Sciences, Harbin 150070, China
cAcademy of Fundamental and Interdisciplinary Sciences, Harbin Institute of Technology, Harbin 150080, China. E-mail: zxd9863@163.com
First published on 28th August 2014
Abstract
A facile, industrially viable strategy is proposed for the scalable production of transition metal disulphide/graphite nanoflake composites by a combination of ball milling and short-time sonication. The experimental conditions are mild and energy efficient, and the yields are fairly high. This strategy can produce larger MoS2 and WS2 nanoflakes with more lithium storage sites than the conventional, long-time sonication method. Besides, the obtained graphite nanoflakes have a higher degree of lattice integrity than reduced graphene oxide that is structurally permanently damaged, and can thus serve as a high-efficiency conductive additive. A prominent synergy is witnessed between the excellent electrochemical performances of the MoS2 and WS2 nanoflakes and the high electronic conductivity of the graphite nanoflakes. The resulting MoS2 and WS2/graphite nanoflake composites exhibit superior lithium storage capacities, cycling stabilities and rate capabilities, thus providing a basis for developing high-performance anodes of next-generation lithium-ion batteries.
1. Introduction
Nowadays the global energy and environmental problems are becoming increasingly obvious and serious. Therefore, how to develop clean and renewable energy sources is an urgent task of today. The lithium-ion battery (LIB), among other candidates, is an ideal driving force for portable electronics due to its high energy density, low self-discharging rate and long service life.1–4 However, the ever-increasing demands for large-size power tools, such as electric vehicles and power grids, set higher standards on the energy density and safety of the secondary power systems. As such, the present anode material of LIBs, i.e., graphitic carbon, is not satisfactory due to its low theoretical capacity (372 mA h g−1). Moreover, the lithium dendrites generated on the surface of the graphitic carbon anodes during the fast charging processes raise serious safety concerns. In this sense, finding suitable anode substitutes with better electrochemical performances is a challenging yet promising competition in which researchers all over the world are involved.Transition metal disulphides, such as MoS2 and WS2, are typical members of a huge layered compound family.5,6 In this layered structure, the transition metal and sulphur atoms are tightly bonded by strong covalent forces to form layers, while the adjacent layers are loosely bound by weak van der Waals forces. When Li+ ions diffuse into the galleries between layers, a redox reaction occurs according to the following mechanism (M = Mo or W):
1st discharge
| MS2 + xLi+ + xe− → LixMS2 | (1) |
| LixMS2 + (4 − x)Li+ + (4 − x)e− → M + 2Li2S | (2) |
1st charge
| 2Li2S ↔ S + 2Li+ + 2e− | (3) |
After exfoliation, the resulting MoS2 and WS2 nanoflakes possess larger gallery spacings and shorter diffusion pathways, which translate into higher lithium storage capacities than the bulk forms. Recently novel anode materials based on MoS2 and WS2 nanoflakes, as promising substitutes for graphitic carbon, have been extensively exploited.7–17 It is worth noting, however, that although MoS2 and WS2 have superior ionic conductivity, their extremely low electronic conductivity inevitably leads to poor cycle and rate performances, which largely hamper their practical applications in next-generation LIBs.
To overcome this embarrassment, researchers are endeavoured to dope MoS2 and WS2 with conductive additives, thereby fabricating various conductive composites to improve their electronic conductivity. Carbonaceous materials, such as amorphous carbon18–21 and carbon nanotubes,22–27 are the most widely employed conductive additives. Very recently attempts have been made on the in situ hydrothermal synthesis of conductive composites of MoS2 or WS2 and reduced graphene oxide (r-GO).28–40 It is found that the cycle stabilities and rate capabilities of the MoS2 or WS2/r-GO composites are enhanced because r-GO can facilitate easy electron transport. However, the rigourous experimental conditions and low yields associated with the in situ hydrothermal synthesis set limitations to the scalable production of the MoS2 or WS2/r-GO composite anodes. To address this issue, an alternative method involving simple physical mixing of pre-synthesised MoS2 or WS2 and r-GO has been employed by ourselves41 and others.42 Whereas, the biggest problem regarding the use of r-GO as a conductive additive lies in its irreversibly damaged structure due to the oxidation–reduction process. The reported electronic conductivity of r-GO varies from 0.17–3.05 S cm−1 (reduced by NaBH4),43,44 20 S cm−1 (reduced by Al powder),45 26.9–77 S cm−1 (reduced by vitamin C),46 to 41–99 S cm−1 (reduced by hydrazine hydrate).46 These values are rather inadequate and non-uniform, making r-GO an inferior conductive additive to graphite nanoflakes that have a perfect hexagonal symmetry.
Based on our experience on the top-down liquid-phase exfoliation of graphite and transition metal disulphide nanoflakes from their bulk forms,47–50 here we propose a facile, industrially viable strategy for the scalable production of MoS2 and WS2/graphite nanoflake composites by a combination of ball milling and short-time sonication. The experimental conditions are mild and energy efficient, and the yields are fairly high compared to the in situ hydrothermal synthesis. This strategy can produce larger MoS2 and WS2 nanoflakes with more lithium storage sites than the conventional, long-time sonication method. Moreover, this strategy can also produce graphite nanoflakes with a higher degree of lattice integrity than r-GO, and their electronic conductivity is up to 163 S cm−1 (tested on a pressed pellet by the standard four-probe technique). The resulting MoS2 and WS2/graphite nanoflake composites exhibit superior lithium storage capacities, cycle stabilities and rate capabilities when evaluated as anode materials of LIBs, laying a basis for the industrial applications of high-performance anodes for next-generation LIBs.
2. Experimental section
2.1. Materials and method
Natural graphite powder (particle sizes ≤ 300 mesh) was purchased from Sinopharm Chemical Reagent Co., Ltd. MoS2 and WS2 powders (particle sizes = ∼6 μm) were purchased from Sigma-Aldrich Co., LLC. N,N-Dimethylformide (DMF) and N-methylpyrrolidone (NMP) were purchased from Beijing Chemical Works. Acetylene black (battery grade) was purchased from Shenzhen Luhua Industrial and Trading Co., Ltd. Poly(vinylidene fluoride) (PVDF, KYNAR 761) was purchased from Arkema Inc.In a typical experiment, natural graphite, MoS2 or WS2 powder 2 g and DMF 200 mL were added to a PA6 vial loaded with ZrO2 balls (6 mm in diameter) 500 g, and milled in a planetary ball mill (KQM-XH, Xianyang Jinhong General Machinery Co., Ltd.) at 300 rpm for 24 h. The slurries were taken out, mixed at different wt ratios, sonicated at 300 W for 30 min, vacuum-filtered, and dried at 100 °C for 24 h. The resulting composites were then annealed at 800 °C in nitrogen for 2 h.
An anode was prepared by coating a copper foil with a slurry containing 70 wt% active material, 10 wt% acetylene black and 20 wt% PVDF dissolved in NMP. The anode was dried at 120 °C in vacuum for 12 h, and equipped in a half cell according to the configuration of (−) Li|electrolyte|anode (+) in a vacuum glove box. The electrolyte is 1 M solution of LiPF6 in ethylene carbonate–dimethyl carbonate at a vol ratio of 1/1. The separator is a microporous polypropylene membrane.
2.2. Sample characterisation
Transmission electron microscopy (TEM) and high-resolution TEM (HRTEM) were performed by a JEOL JEM-2010 microscope operated at an accelerating voltage of 120 kV.Scanning electron microscopy (SEM) was performed by a JEOL JSM-7401F microscope operated at an accelerating voltage of 3.0 kV.
Scanning probe microscopy (SPM) was performed by a Shimadzu SPM-9700 microscope operated in the tapping mode.
Raman spectroscopy was performed by a Renishaw RM2000 spectrometer (λ = 514 nm).
X-ray diffraction (XRD) spectroscopy was performed by a PANalytical X'Pert PRO spectrometer with Cu Kα radiation (λ = 1.5418 Å).
UV/vis spectroscopy was performed by a Persee TU-1810 spectrometer.
Brunauer–Emmett–Teller (BET) measurement was performed by a Quantachrome Autosorb-1 Canalyser (USA).
3. Results and discussion
The combination of ball milling and short-time sonication can realise the scalable production of high-quality nanoflakes at relatively low energy and chemicals consumption. Recently, several groups have reported the successful application of ball milling for exfoliating layered materials in different solvents.51–55 The difference lies in the rotary speed and time, which are key to the morphology and size of the final product. In our case, the ball milling process is aimed at the preliminary thinning of the bulk powders, and the subsequent short-time sonication process is devoted to further breaking them into large-area nanoflakes. Compared to the conventional, long-time sonication method, this strategy is industrially viable since the experimental conditions are mild and energy efficient, and the yields are fairly high. A morphological observation of the resulting nanoflakes is realised by TEM characterisation (Fig. 1). The nanoflakes shown in the TEM images have large lateral sizes of 2–5 μm, an advantage over long-time sonication that often produces small pieces.5,47–50 As seen from the corresponding electron diffraction (ED) patterns, these nanoflakes are highly crystalline, suggesting a high degree of lattice integrity. The HRTEM images reveal that a substantial fraction of the obtained nanoflakes are ≤10 layers. The measured gallery spacings of ∼0.33, 0.65 and 0.64 nm are in good agreement with the (002) values reported for graphite, MoS2 and WS2, respectively.6,56 The atomically resolved HRTEM images confirm that the honeycomb structure of the nanoflakes is largely preserved with few, if any, disorders (Fig. S7–S9†). The nature of the nanoflakes can be further clarified by AFM analysis, as shown in Fig. S10.† It can be seen that the nanoflakes have a wide size distribution ranging from hundreds of nanometres to several micrometres. Note that most of the nanoflakes, even those whose lateral sizes are on the micrometre scale, have thicknesses ≤10 nm, which support the HRTEM results. These large, thin MoS2 and WS2 nanoflakes are believed to be able to provide more lithium storage sites than the small ones (generally <100 nm) produced by conventional, long-time sonication method.5 | ||
| Fig. 1 TEM images of (a) graphite, (c) MoS2 and (e) WS2 nanoflakes and the corresponding ED patterns (insert); HRTEM images of (b) graphite, (d) MoS2 and (f) WS2 nanoflakes. | ||
Raman spectroscopy can provide an insight into the crystalline nature of the nanoflakes and their composites. Fig. 2 shows the Raman spectra of these composites at different wt ratios. As seen from the figure, the Raman spectrum of the MoS2 or WS2 nanoflakes is featured by two sharp characteristic peaks, i.e., one at 378.2 or 351.1 cm−1 corresponding to the in-plane E2g vibration, and the other at 403.8 or 415.3 cm−1 corresponding to the out-of-plane A1g vibration.6 On the other hand, the graphite nanoflakes have two well-known characteristic peaks, i.e., peak A1g at 1354.0 cm−1 (also known as peak D) related to the symmetric vibration of the six-fold aromatic rings arising from the structural disorders, and peak E2g at 1580.9 cm−1 (also known as peak G) assigned to the symmetric vibration of the sp2-hybridized carbon atoms in the graphitic sheets.47 The integral intensity ratio of peak D to G, ID/IG, is used as a measure of the structural disorders in a carbon product, that is, the smaller the ID/IG value, the better the lattice integrity. In our experiment, the ID/IG value of the graphite nanoflakes is only 0.185, which is significantly lower than that of r-GO,41 indicating the former suffer from much fewer structural disorders and thus reasonably exhibit a much higher electronic conductivity (163 S cm−1). The Raman spectra of the graphite powder and nanoflakes are presented in Fig. S11.† The prominent peak occurring at 2708.6 cm−1, i.e., peak 2D, is the second order of peak D. The shape of peak 2D, an important indicator of the number of layers, has been systematically studied for the graphite powder or graphene with various layers, and found to be considerably different from each other.57–59 When the number of layers is >10, the corresponding Raman spectrum is almost identical to that of the graphite powder. A careful comparison informs us that the Raman spectrum of our graphite nanoflakes, unlike that of the graphite powder, is in consistent with those of 5–10 layers, verifying the successful exfoliation of the graphite powder into few-layer nanoflakes. The Raman result is in good agreement with the HRTEM and AFM data. As to the composites, the characteristic peaks of both components (MoS2 or WS2 and graphite nanoflakes) can be clearly seen, confirming that the physical mixing process effectively preserves the crystalline nature without introducing any new damage.
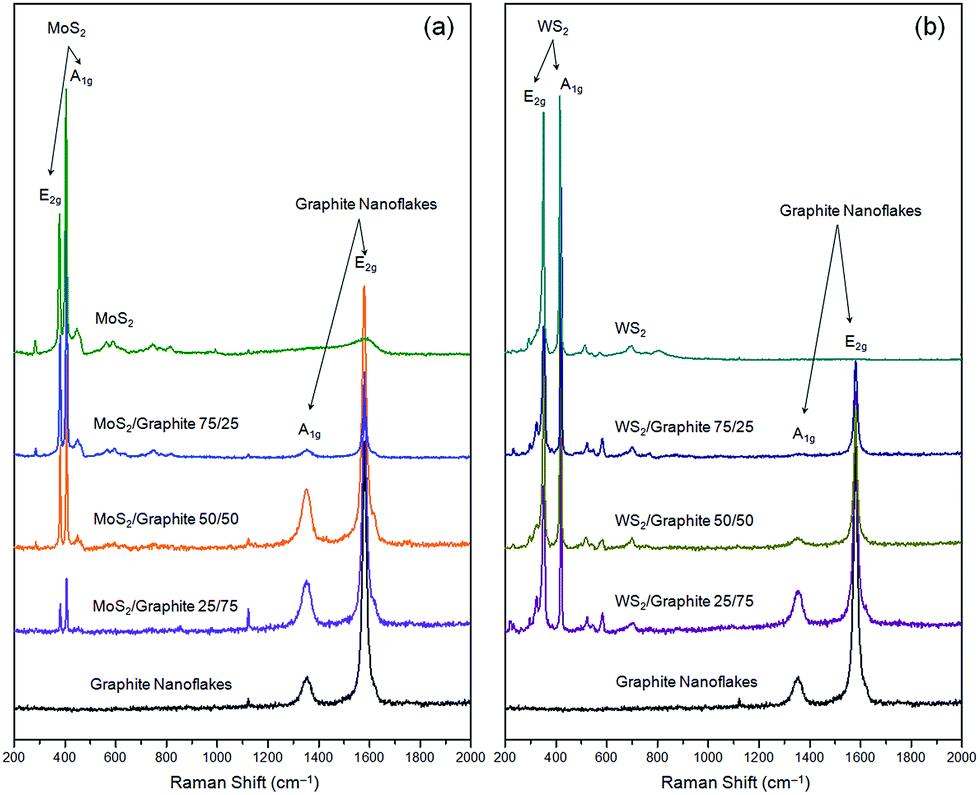 | ||
| Fig. 2 Raman spectra of (a) MoS2/graphite and (b) WS2/graphite nanoflake composites at different wt ratios. | ||
To understand the morphological features of the MoS2 and WS2/graphite nanoflake composites, we present two typical SEM images in Fig. 3. It is clearly seen that the lateral sizes of the nanoflakes range from hundreds of nanometres to several micrometres, statistically larger than the ones produced by conventional, long-time sonication that often results in the breakage of the nanoflakes into small pieces due to the strong shearing forces.5,47–50 It is hard to distinguish the MoS2 or WS2 nanoflakes from the graphite nanoflakes due to their similar morphology.42 A closer observation on the edges of the nanoflakes shows that the thicknesses are around 10 nm or below, implying that the nanoflakes are mostly few-layer ones. It is worth noting that the nanosheets are uniformly mixed and loosely packed without obvious restacking, ensuring sufficient pores and voids that are desirable for creating pathways for Li+ ion and electron diffusion, and are also desirable for buffering the volume expansion during the repeated charging–discharging processes. Therefore, these MoS2 and WS2/graphite nanoflake composites are expected to exhibit excellent electrochemical performances as will be proven below. The uniform mixing of the nanoflakes can be further verified by HRTEM characterisation, as shown in Fig. S14.† The edges of the MoS2 and graphite nanoflakes can be clearly differentiated as indicated by arrows.
 | ||
| Fig. 3 Typical SEM images of (a) MoS2/graphite and (b) WS2/graphite nanoflake composites at a wt ratio of 50/50. | ||
The XRD patterns of the MoS2 and WS2/graphite nanoflake composites at different wt ratios are presented in Fig. 4. The XRD patterns of the MoS2 and WS2 nanoflakes show a series of diffraction peaks as resolved in the figure (JCPDS card nos 37-1492 and 84-1398), which confirm the highly crystalline nature of the liquid-exfoliated MoS2 and WS2 nanoflakes compared to the ion-intercalated6 or hydrothermally synthesised ones.28–40 The primary (002) diffraction peaks reside at 14.42° (MoS2) and 14.37° (WS2), corresponding to d-spacings of 0.613 and 0.616 nm, respectively. These values are in good agreement with HRTEM observation (Fig. 1d and f), confirming that the nanoflakes exist mainly in the form of few layers. The XRD pattern of the graphite nanoflakes has a primary (002) diffraction peak at 26.58°, which represents a d-spacing of 0.335 nm.60 This value is also in good agreement with HRTEM observation (Fig. 1b). Note that the crystallinity of the graphite nanoflakes is much higher than that of r-GO as revealed by XRD characterisation, which reasonably explains the better structural integrity and higher electronic conductivity of the former. After the liquid-phase mixing, the resulting MoS2 and WS2/graphite nanoflake composites do not show obvious differences in terms of peak shape and position, once again confirming that the physical mixing process does not cause structural disorders.
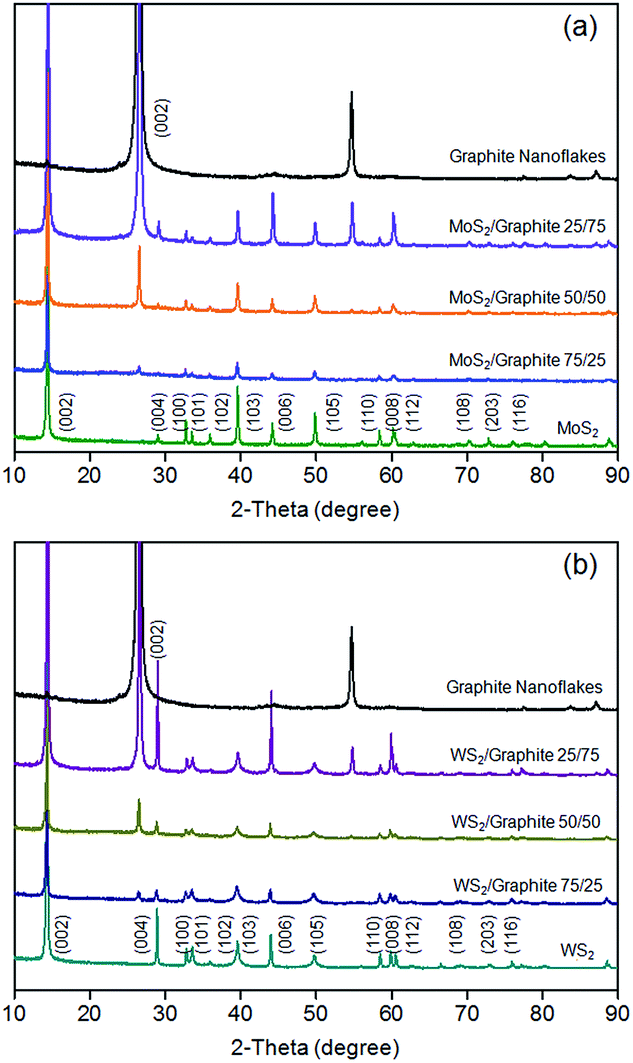 | ||
| Fig. 4 XRD patterns of (a) MoS2/graphite and (b) WS2/graphite nanoflake composites at different wt ratios. | ||
The BET data can provide us with sufficient information on the specific surface areas and pore volumes of MoS2, WS2 and graphite nanoflakes as well as their composites, which are summarized in Table 1. As discussed above, the combination of ball milling and short-time sonication can produce “gigantic” MoS2, WS2 and graphite nanoflakes with relatively large specific surface areas of 168.89, 56.16 and 25.08 m2 g−1, respectively. Introducing large-area graphite nanoflakes can thus increase the specific surface areas of the composites, and prevent the MoS2 and WS2 nanoflakes from restacking. Therefore after the liquid-phase mixing, the 50/50 MoS2 and WS2/graphite nanoflake composites have specific surface areas up to 78.81 and 36.74 m2 g−1 (∼1.5 times increase), which are much larger than the value reported for the conventional, long-time sonication method,42 and are advantageous for Li+ ion and electron storage. The larger specific surface area of the MoS2/graphite nanoflake composite than its WS2 counterpart may partially explain the better electrochemical performances of the former, as will be proven below. Moreover, the pore volumes of the MoS2 and WS2/graphite nanoflake composites, obtained by Barrett–Joyner–Halenda (BJH) calculation, are 0.12 and 0.07 cm3 g−1, respectively. The relatively large pore volumes, mainly contributed by the large-area, flexible graphite nanoflakes, are desirable for facilitating the electrolyte infiltration as well as buffering the volume expansion during the repeated charging–discharging processes.
| Sample name | Specific surface area (m2 g−1) | Pore volume (cm3 g−1) |
|---|---|---|
| MoS2 nanoflakes | 56.16 | 0.07 |
| WS2 nanoflakes | 25.08 | 0.06 |
| Graphite nanoflakes | 168.89 | 0.21 |
| MoS2/graphite 50/50 | 78.81 | 0.12 |
| WS2/graphite 50/50 | 36.74 | 0.07 |
Fig. 5 shows the first three charging–discharging and cyclic voltammetry (CV) curves of two representative composites, i.e., the 50/50 MoS2 and WS2/graphite nanoflake composites. As seen from Fig. 5a, the first discharging process of the MoS2/graphite nanoflake composite is featured by three plateaus at ∼1.1, 0.5 and 0.1 V, indicating the formation of LixMoS2, the conversion of Mo4+ to Mo nanoparticles (embedded in a Li2S matrix), and the intercalation of Li+ ions into the galleries of the graphite nanoflakes (Fig. S15†), respectively. The slope below ∼0.5 V is generally ascribed to the formation of a solid-electrolyte interface (SEI) layer originating from the electrochemically driven electrolyte decomposition. During the second discharging process, a new plateau at ∼2.2 V occurs, corresponding to a reaction of Mo + 4Li+ + 2S + 4e− → 2Li2S + Mo (Mo is left to emphasise its inertness). Note that the actual polarization between discharge and charge of the MoS2/graphite nanoflake composite is ∼1.7 V, which is too high for the oxidation of Mo to MoS2 (polarization = ∼0.6 V).61 In this sense, the plateaus at ∼0.2 and 2.3 V during the first charging process should be attributed to the extraction of Li+ ions from the galleries of the graphite nanoflakes (Fig. S15†) and the conversion of Li2S to S,8,14 respectively. During the second charging process, a new, inconspicuous plateau at ∼1.7 V occurs, a proof of the partial oxidation of Mo to MoS2. In the case of the WS2/graphite nanoflake composite (Fig. 5c), the lithium storage mechanism is very similar. Basically, during the first discharging process the inconspicuous plateau at ∼1.5 V is attributed to the intercalation of Li+ ions and the formation of LixWS2; the remarkable plateaus at ∼0.7 and 0.1 V are related to a reaction of WS2 + 4Li+ + 4e− → W + 2Li2S (accompanied by the irreversible electrolyte decomposition), and the insertion of Li+ ions into the galleries of the graphite nanoflakes, respectively. During the subsequent discharging process two plateaus occur at ∼1.9 and 2.2 V, which are ascribed to the formation of a gel-like polymeric layer out of the dissolution of Li2S in the electrolyte.39 During the first charging process, the plateaus at ∼2.3 and 0.2 V correspond to the extraction of Li+ ions, which remain unchanged during the subsequent charging process. The charging–discharging results are in good agreement with the CV curves (Fig. 5b and d). Note that the charging–discharging and CV curves of the second and third cycles are nearly superposed, indicating high cycle stabilities of the MoS2 and WS2/graphite nanoflake composites.
 | ||
| Fig. 5 The first three charging–discharging (current density = 100 mA g−1) and CV (scan rate = 0.1 mV s−1) curves of (a and b) MoS2/graphite and (c and d) WS2/graphite nanoflake composites at a wt ratio of 50/50. | ||
Fig. 6 compares the cycle behaviours of MoS2, WS2 and their composites with the graphite nanoflakes. The initial discharge capacities of MoS2 and WS2 are 1223 and 830 mA h g−1, much higher than the theoretical values of their bulk forms because of the electrochemically driven electrolyte decomposition and the enlarged gallery spacings due to exfoliation. However, the capacities fade rapidly to only 337 and 276 mA h g−1 at the 50th cycle, corresponding to retention rates of only 27.5% and 33.3%, respectively. The drastic capacity losses can be ascribed to the poor intrinsic electronic conductivity of transition metal disulphides. After being conductively doped, MoS2 and WS2 have significantly enhanced electrochemical performances. In addition, the large-area graphite nanoflakes can also increase the specific surface areas (Table 1), as well as prevent the MoS2 and WS2 nanoflakes from restacking, which are advantageous for the Li+ ion and electron storage. The optimum compositions are the 50/50 MoS2 and WS2/graphite nanoflake composites, whose reversible capacities are as high as 951 and 782 mA h g−1 after 50 cycles. Note that our MoS2/graphite nanoflake composite even outperforms the CVD-grown MoS2/pristine graphene composite (877 mA h g−1 @ 100 mA g−1) in terms of capacity,62 thus exhibiting more splendid industrial potentials especially when one considers the mild experimental conditions and high yields of our strategy compared to CVD. The 75/25 MoS2 and WS2/graphite nanoflake composites have poorer cycle stabilities, fading from 1186 and 1019 mA h g−1 to 663 and 621 mA h g−1 after 50 cycles, which can be ascribed to the lower content of the graphite nanoflakes leading to more difficult electron transport. The 25/75 MoS2 and WS2/graphite nanoflake composites show fairly good cycle performances, while their lower reversible capacities (538 and 485 mA h g−1) can be assigned to the higher content of the graphite nanoflakes whose lithium storage properties are inferior to MoS2 and WS2. It is shown that the reversible capacity of the graphite nanoflakes is only 267 mA h g−1 after 50 cycles (Fig. S16†), which is comparable to the previous reports.63,64
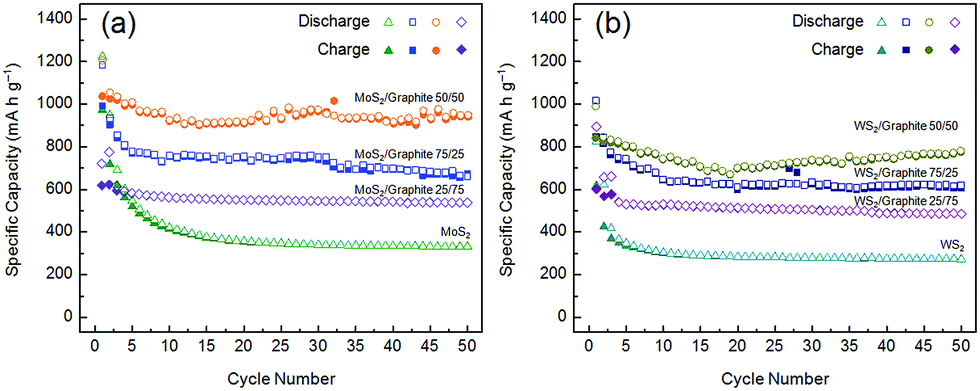 | ||
| Fig. 6 Cycle behaviours (current density = 100 mA g−1) of (a) MoS2/graphite and (b) WS2/graphite nanoflake composites at different wt ratios. | ||
The rate capabilities of the 50/50 MoS2 and WS2/graphite nanoflake composites are also evaluated, as shown in Fig. 7. When the current density is raised to 500 mA g−1, the reversible capacity of the MoS2/graphite nanoflake composite is above 800 mA h g−1, still better than that of the CVD-grown MoS2/pristine graphene composite (665 mA h g−1 @ 500 mA g−1).62 Even at a very high current density of 2000 mA g−1, the reversible capacity is still 625 mA h g−1. Upon the recovery of the current density to 100 mA g−1, the reversible capacity returns to 938 mA h g−1, further proving the high cyclability of the MoS2/graphite nanoflake composite. As to the WS2/graphite nanoflake composite, the reversible capacities are 508, 492 and 488 mA h g−1 at current densities of 500, 1000 and 2000 mA g−1, respectively. The tremendous enhancement in the rate capabilities also demonstrates the effectiveness of introducing the graphite nanoflakes as a high-efficiency conductive additive for transition metal disulphides. To elucidate the effect of the graphite nanoflakes, we have conducted electrochemical impedance spectroscopy (EIS) on MoS2, WS2 and their composites with the graphite nanoflakes, and the results are shown in Fig. S17 and S18.† The diameters of the high- and medium-frequency semicircles for MoS2 or WS2, corresponding to the SEI film resistance (Rf) of 16.5 or 20.2 Ω and the charge-transfer resistance (Rct) of 57.1 or 109.2 Ω, are significantly reduced to 9.7 or 18.1 Ω and 15.5 or 13.6 Ω after being conductively doped with the graphite nanoflakes. The partial electron transfer from graphite to the transition metal disulphide layers has been demonstrated both theoretically and experimentally.65 It is inferred that the uniform mixing of the MoS2 or WS2 and graphite nanoflakes can largely increase the electronic conductivity, which facilitates easy electron transport from the current collector to the electrode. Therefore, the cycle and rate performances of the MoS2 and WS2/graphite nanoflake composites are significantly enhanced compared to those of neat MoS2 and WS2.
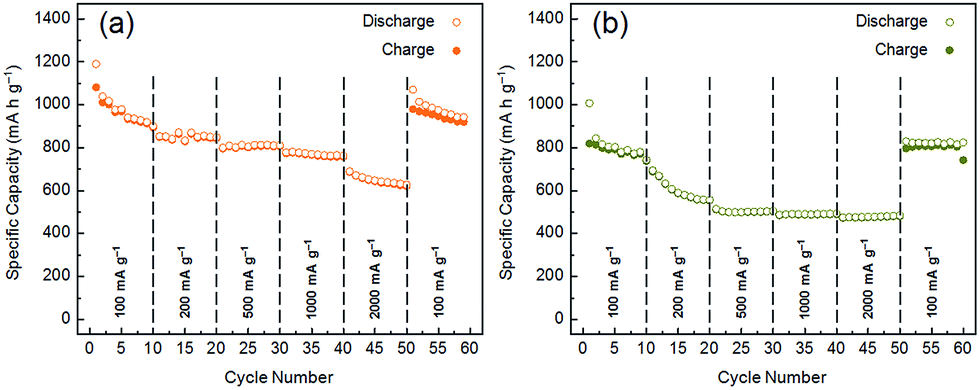 | ||
| Fig. 7 Rate capabilities of (a) MoS2/graphite and (b) WS2/graphite nanoflake composites at a wt ratio of 50/50. | ||
4. Conclusions
In summary, we have successfully proposed a facile, industrially viable strategy for the scalable production of MoS2 and WS2/graphite nanoflake composites by a combination of ball milling and short-time sonication. This strategy can produce larger MoS2 and WS2 nanoflakes with more lithium storage sites than the conventional, long-time sonication method. Besides, the obtained graphite nanoflakes have a higher degree of lattice integrity than r-GO that is structurally permanently damaged, and can thus serve as a high-efficiency conductive additive. A prominent synergy between the excellent electrochemical performances of the MoS2 and WS2 nanoflakes and the high electronic conductivity of the graphite nanoflakes is witnessed. The MoS2 and WS2/graphite nanoflake composites exhibit superior lithium storage capacities, cycle stabilities and rate capabilities, thus laying a basis for developing high-performance anodes for next-generation LIBs.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (nos 21304053 and 21274079) and the Specialized Research Fund for the Doctoral Program of Higher Education (no. 20120002130012). Y.-T. L. is grateful to the China Postdoctoral Science Foundation (no. 2014T70077). X.-D. Z. is grateful to the Postdoctoral Science-Research Development Foundation of Heilongjiang Province (no. LBH-Q11130) and Natural Science Foundation of Heilongjiang Province (no. B201202)..Notes and references
- Y. Deng, L. Wan, Y. Xie, X. Qin and G. Chen, RSC Adv., 2014, 4, 23914 RSC
.
- D. Kong, H. He, Q. Song, B. Wang, Q.-H. Yang and L. Zhi, RSC Adv., 2014, 4, 23372 RSC
- H. Wang, J. Liu, X. Wang, C. Wu, Q. Zhao, Y. Fu, X. Yang and H. Shu, RSC Adv., 2014, 4, 22241 RSC
- L. Qiu, Z. Shao, W. Wang, F. Wang, D. Wang, Z. Zhou, P. Xiang and C. Xu, RSC Adv., 2014, 4, 24859 RSC
- J. N. Coleman, M. Lotya, A. O'Neill, S. D. Bergin, P. J. King, U. Khan, K. Young, A. Gaucher, S. De, R. J. Smith, I. V. Shvets, S. K. Arora, G. Stanton, H.-Y. Kim, K. Lee, G. T. Kim, G. S. Duesberg, T. Hallam, J. J. Boland, J. J. Wang, J. F. Donegan, J. C. Grunlan, G. Moriarty, A. Shmeliov, R. J. Nicholls, J. M. Perkins, E. M. Grieveson, K. Theuwissen, D. W. McComb, P. D. Nellist and V. Nicolosi, Science, 2011, 331, 568 CrossRef CAS PubMed
- H. S. S. Ramakrishna Matte, A. Gomathi, A. K. Manna, D. J. Late, R. Datta, S. K. Pati and C. N. R. Rao, Angew. Chem., Int. Ed., 2010, 49, 4059 CrossRef PubMed
- G. Du, Z. Guo, S. Wang, R. Zeng, Z. Chen and H. Liu, Chem. Commun., 2010, 46, 1106 RSC
- X. Fang, C. Hua, X. Guo, Y. Hu, Z. Wang, X. Gao, F. Wu, J. Wang and L. Chen, Electrochim. Acta, 2012, 81, 155 CrossRef CAS PubMed
- S. Ding, D. Zhang, J. S. Chen and X. W. Lou, Nanoscale, 2012, 4, 95 RSC
- Y. Li, D. Wu, Z. Zhou, C. R. Cabrera and Z. Chen, J. Phys. Chem. Lett., 2012, 3, 2221 CrossRef CAS
- S.-K. Park, S.-H. Yu, S. Woo, J. Ha, J. Shin, Y.-E. Sung and Y. Piao, CrystEngComm, 2012, 14, 8323 RSC
- C. Feng, J. Ma, H. Li, R. Zeng, Z. Guo and H. Liu, Mater. Res. Bull., 2009, 44, 1811 CrossRef CAS PubMed
- H. Liu, D. Su, G. Wang and S. Z. Qiao, J. Mater. Chem., 2012, 22, 17437 RSC
- X. Fang, C. Hua, C. Wu, X. Wang, L. Shen, Q. Kong, J. Wang, Y. Hu, Z. Wang and L. Chen, Chem.–Eur. J., 2013, 19, 5694 CrossRef CAS PubMed
- R. Bhandavat, L. David and G. Singh, J. Phys. Chem. Lett., 2012, 3, 1523 CrossRef CAS
- C. Feng, L. Huang, Z. Guo and H. Liu, Electrochem. Commun., 2007, 9, 119 CrossRef CAS PubMed
- L. Zhang, H. B. Wu, Y. Yan, X. Wang and X. W. Lou, Energy Environ. Sci., 2014 10.1039/C4EE01932F
- C. Zhang, Z. Wang, Z. Guo and X. W. Lou, ACS Appl. Mater. Interfaces, 2012, 4, 3765 CAS
- C. Zhang, H. B. Wu, Z. Guo and X. W. Lou, Electrochem. Commun., 2012, 20, 7 CrossRef CAS PubMed
- K. Chang, W. Chen, L. Ma, H. Li, H. Li, F. Huang, Z. Xu, Q. Zhang and J.-Y. Lee, J. Mater. Chem., 2011, 21, 6251 RSC
- L. Zhang and X. W. Lou, Chem.–Eur. J., 2014, 20, 5219 CrossRef CAS PubMed
- K. Bindumadhavan, S. K. Srivastava and S. Mahanty, Chem. Commun., 2013, 49, 1823 RSC
- S.-Y. Tai, C.-J. Liu, S.-W. Chou, F. S.-S. Chien, J.-Y. Lin and T.-W. Lin, J. Mater. Chem., 2012, 22, 24753 RSC
- S.-K. Park, S.-H. Yu, S. Woo, B. Quan, D.-C. Lee, M. K. Kim, Y.-E. Sung and Y. Piao, Dalton Trans., 2013, 42, 2399 RSC
- S. Ding, J. S. Chen and X. W. Lou, Chem.–Eur. J., 2011, 17, 13142 CrossRef CAS PubMed
- J.-Z. Wang, L. Lu, M. Lotya, J. N. Coleman, S.-L. Chou, H.-K. Liu, A. I. Minett and J. Chen, Adv. Energy Mater., 2013, 3, 798 CrossRef CAS PubMed
- B. Hu, X. Qin, A. M. Asiri, K. A. Alamry, A. O. Al-Youbi and X. Sun, Electrochem. Commun., 2013, 28, 75 CrossRef CAS PubMed
- X. Zhou, Z. Wang, W. Chen, L. Ma, D. Chen and J. Y. Lee, J. Power Sources, 2014, 251, 264 CrossRef CAS PubMed
- X. Jiang, X. Yang, Y. Zhu, J. Shen, K. Fan and C. Li, J. Power Sources, 2013, 237, 178 CrossRef CAS PubMed
- K. Chang and W. Chen, Chem. Commun., 2011, 47, 4252 RSC
- K. Chang and W. Chen, ACS Nano, 2011, 5, 4720 CrossRef CAS PubMed
- Z. Wang, T. Chen, W. Chen, K. Chang, L. Ma, G. Huang, D. Chen and J. Y. Lee, J. Mater. Chem. A, 2013, 1, 2202 CAS
- G. Huang, T. Chen, W. Chen, Z. Wang, K. Chang, L. Ma, F. Huang, D. Chen and J. Y. Lee, Small, 2013, 9, 3693 CrossRef CAS PubMed
- Y. Gong, S. Yang, Z. Liu, L. Ma, R. Vajtai and P. M. Ajayan, Adv. Mater., 2013, 25, 3979 CrossRef CAS PubMed
- K. Chang and W. Chen, J. Mater. Chem., 2011, 21, 17175 RSC
- X. Zhou, L.-J. Wan and Y.-G. Guo, Chem. Commun., 2013, 49, 1838 RSC
- H. Yu, C. Ma, B. Ge, Y. Chen, Z. Xu, C. Zhu, C. Li, Q. Ouyang, P. Gao, J. Li, C. Sun, L. Qi, Y. Wang and F. Li, Chem.–Eur. J., 2013, 19, 5818 CrossRef CAS PubMed
- K. Chang, D. Geng, X. Li, J. Yang, Y. Tang, M. Cai, R. Li and X. Sun, Adv. Energy Mater., 2013, 3, 839 CrossRef CAS PubMed
- K. Shiva, H. S. S. Ramakrishna Matte, H. B. Rajendra, A. J. Bhattacharyya and C. N. R. Rao, Nano Energy, 2013, 2, 787 CrossRef CAS PubMed
- D. Chen, G. Ji, B. Ding, Y. Ma, B. Qu, W. Chen and J. Y. Lee, Nanoscale, 2013, 5, 7890 RSC
- Y.-T. Liu, X.-D. Zhu, Z.-Q. Duan and X.-M. Xie, Chem. Commun., 2013, 49, 10305 RSC
- V. H. Pham, K.-H. Kim, D.-W. Jung, K. Singh, E.-S. Oh and J. S. Chung, J. Power Sources, 2013, 244, 280 CrossRef CAS PubMed
- H.-J. Shin, K. K. Kim, A. Benayad, S.-M. Yoon, H. K. Park, I.-S. Jung, M. H. Jin, H.-K. Jeong, J. M. Kim, J.-Y. Choi and Y. H. Lee, Adv. Funct. Mater., 2009, 19, 1987 CrossRef CAS PubMed
- Y. Liang, D. Wu, X. Feng and K. Müllen, Adv. Mater., 2009, 21, 1679 CrossRef CAS PubMed
- Z. Fan, K. Wang, T. Wei, J. Yan, L. Song and B. Shao, Carbon, 2010, 48, 1686 CrossRef CAS PubMed
- M. J. Fernández-Merino, L. Guardia, J. I. Paredes, S. Villar-Rodil, P. Solís-Fernández, A. Martínez-Alonso and J. M. D. Tascón, J. Phys. Chem. C, 2010, 114, 6426 Search PubMed
- Y.-T. Liu, X.-M. Xie and X.-Y. Ye, Carbon, 2011, 49, 3529 CrossRef CAS PubMed
- Y.-T. Liu, Z. Tan, X.-M. Xie, Z.-F. Wang and X.-Y. Ye, Chem.–Asian J., 2013, 8, 817 CrossRef CAS PubMed
- L. Pan, Y.-T. Liu, X.-M. Xie and X.-D. Zhu, Chem.–Asian J., 2014, 9, 1519 CrossRef CAS PubMed
- Y.-T. Liu, Z.-Q. Duan, X.-M. Xie and X.-Y. Ye, Chem. Commun., 2013, 49, 1642 RSC
- Y. Yao, Z. Lin, Z. Li, X. Song, K.-S. Moon and C.-P. Wong, J. Mater. Chem., 2012, 22, 13494 RSC
- K. R. Paton, E. Varrla, C. Backes, R. J. Smith, U. Khan, A. O'Neill, C. Boland, M. Lotya, O. M. Istrate, P. King, T. Higgins, S. Barwich, P. May, P. Puczkarski, I. Ahmed, M. Moebius, H. Pettersson, E. Long, J. Coelho, S. E. O'Brien, E. K. McGuire, B. M. Sanchez, G. S. Duesberg, N. McEvoy, T. J. Pennycook, C. Downing, A. Crossley, V. Nicolosi and J. N. Coleman, Nat. Mater., 2014, 13, 624 CrossRef CAS PubMed
- C. Knieke, A. Berger, M. Voigt, R. N. Klupp Taylor, J. Röhrl and W. Peukert, Carbon, 2010, 48, 3196 CrossRef CAS PubMed
- Y. Yao, L. Tolentino, Z. Yang, X. Song, W. Zhang, Y. Chen and C.-P. Wong, Adv. Funct. Mater., 2013, 23, 3577 CrossRef CAS PubMed
- C. Damm, J. Körner and W. Peukert, J. Nanopart. Res., 2013, 15, 1561 CrossRef
- E. J. Yoo, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277 CrossRef CAS PubMed
- A. C. Ferrari, J. C. Meyer, V. Scardaci, C. Casiraghi, M. Lazzeri, F. Mauri, S. Piscanec, D. Jiang, K. S. Novoselov, S. Roth and A. K. Geim, Phys. Rev. Lett., 2006, 97, 187401 CrossRef CAS
- L. M. Malard, M. A. Pimenta, G. Dresselhaus and M. S. Dresselhaus, Phys. Rep., 2009, 473, 51 CrossRef CAS PubMed
- U. Khan, A. O'Neill, M. Lotya, S. De and J. N. Coleman, Small, 2010, 6, 864 CrossRef CAS PubMed
- A. Malesevic, R. Vitchev, K. Schouteden, A. Volodin, L. Zhang, G. Van Tendeloo, A. Vanhulsel and C. Van Haesendonck, Nanotechnology, 2008, 19, 305604 CrossRef PubMed
- Y. Kim and J. B. Goodenough, J. Phys. Chem. C, 2008, 112, 15060 CAS
- X. Cao, Y. Shi, W. Shi, X. Rui, Q. Yan, J. Kong and H. Zhang, Small, 2013, 9, 3433 CrossRef CAS PubMed
- S.-L. Chou, J.-Z. Wang, M. Choucair, H.-K. Liu, J. A. Stride and S.-X. Dou, Electrochem. Commun., 2010, 12, 303 CrossRef CAS PubMed
- E. Yoo, J. Kim, E. Hosono, H. Zhou, T. Kudo and I. Honma, Nano Lett., 2008, 8, 2277 CrossRef CAS PubMed
- B. Sachs, L. Britnell, T. O. Wehling, A. Eckmann, R. Jalil, B. D. Belle, A. I. Lichtenstein, M. I. Katsnelson and K. S. Novoselov, Appl. Phys. Lett., 2013, 103, 251607 CrossRef PubMed
Footnotes |
| † Electronic supplementary information (ESI) available: Supplementary figures of the exfoliated 2D nanosheets. See DOI: 10.1039/c4ra05640j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2014 |