One-pot synthesis of porous magnetic cellulose beads for the removal of metal ions†
Xiaolin Yua,
Dongjuan Kanga,
Yanyong Hua,
Shengrui Tong*a,
Maofa Ge*a,
Changyan Caob and
Weiguo Songb
aState Key Laboratory for Structural Chemistry of Unstable and Stable Species, Beijing National Laboratory for Molecular Sciences (BNLMS), Institute of Chemistry, Chinese Academy of Sciences, Beijing, 100190, P. R. China. E-mail: gemaofa@iccas.ac.cn; tongsr@iccas.ac.cn
bLaboratory for Molecular Nanostructures and Nanotechnology, Institute of Chemistry, Chinese Academy of Sciences, Beijing 100190, P. R. China
First published on 4th July 2014
Abstract
Porous magnetic cellulose beads have been successfully prepared via a facile one-pot wet extrusion by in situ depositing CoFe2O4 nanoparticles on cellulose beads. Cellulose beads serve as a template to prevent CoFe2O4 particles from aggregating. The deposited CoFe2O4 nanoparticles are tightly entrapped within cellulose beads because of the strong attractive interactions between the cellulose beads and CoFe2O4 particles. The functionalized cellulose beads can be easily separated from aqueous solution by the external magnetic field. The encapsulated CoFe2O4 nanoparticles act as the magnetic substrate and the active sites to adsorb metal ions. An attractive feature of this preparation method is that it is versatile to prepare a variety of cellulose-based functional nanocomposites in the form of macroscopic beads by incorporating functional particles into the pores of cellulose beads.
1. Introduction
Inorganic/organic hybrid composites with multiple functions have been attracting more and more attention in recent years due to their outstanding mechanical, optical, catalytic and electrical properties.1–3 Magnetic nanomaterials are a class of important functional material because of their high efficiency, cost effectiveness and rapidity of magnetic separation, which have been extensively used for various applications, including gas sensors, electrodes, drug delivery and water treatment.4–6 However, magnetic nanomaterials often generate irreversible aggregation, resulting in the decrease of particle reactivity and specific surface area. Thus, it is extremely important to prevent nanoparticles from aggregating during the fabrication process. An attractive approach that solves the problem of aggregation is to stabilize and functionalize the particles by using organic materials as scaffolds for inorganic crystal growth. The inorganic particles coated by an organic template are considerably more stable against aggregation owing to a large decrease of their surface energy compared with bare particles.7Cellulose is one of the most abundant, natural and renewable biopolymer,8 which is considered to be a promising organic candidate for fabrication of inorganic/organic hybrid composites. This is due to the fact that cellulose can be adsorbed strongly onto the inorganic particles surface by strong van der Waals attractive interaction between the surface and the numerous hydroxyl groups in the repeating glucose units of the cellulose.4,7 However, so far, many research focus on the ex situ approach to fabricate the hybrid composites, where the inorganic nanoparticles are first synthesized in one way, and then are dispersed into an organic solution. Liu et al.9 have reported the synthesis of biocompatible magnetic cellulose–chitosan hybrid gel microspheres reconstituted from ionic liquids via ex situ approach. Luo et al.10 have synthesized the maghemite by coprecipitation of ferrous and ferric chlorides in an ammonium hydroxide solution at first, and then dispersed them into cellulose solution for the production of magnetic cellulose porous microspheres. Although ex situ approach has no limitation for the kinds of inorganic particles and organic materials, a significant disadvantage of this approach is that the preparation procedure is much more complicated compared with in situ approach. Our group has synthesized the cellulose@Fe2O3 hybrid composites by a simple one-pot co-precipitation method.11 However, the potential risk is that it is possible to separate some Fe2O3 nanoparticles from the bulk composites. Thus, there is still a great challenge in developing a facile and efficient method to fabricate various functional inorganic/organic hybrid composites.
Nowadays, water pollution has become a worldwide environmental problem that has serious harm to the human health. The rendering technique for water pollution has evolved into a major research focus. Various materials such as iron oxide,12,13 reduced grapheme oxide,14,15 copper oxide,16 magnesium oxide,17–19 carbon nanotubes,20,21 hydroxyapatite,22,23 cellulose,24–26 have been successfully fabricated to remove pollutants from water. Amongst these materials, iron oxide nanostructures with various morphologies are more effective in the removal of pollutants and have been widely investigated.27–31 However, there is a great risk that the sheet-like nanostructures of iron oxide on the edge of bulk are probably destroyed in the process of using, especially for the test of a fixed bed column that the flow rate of water is rapid. As a result, the destroyed nanosheets difficultly separated from the water will be infused into the water. The introduction of these insoluble nanoscale particles into drinking water can give rise to the potential adverse effects on human health – that is nanotoxicity.32–34
Hence, we demonstrated a facile, one-pot, “green” and efficient method that exploits a versatile system to encapsulate the inorganic particles into the cellulose matrix for fabricating inorganic/organic hybrid cellulose beads with porous structure. The porous cellulose beads can serve as template to prevent the inner particles from aggregating and dissolving when it is used. The particles were dispersed in cellulose beads and more stable against aggregation. Due to the advantage of the particles, the porous cellulose beads has no collapse, thus maintaining the better macroscopic shape, and exhibiting the excellent capability for the removal of the pollutants. Importantly, this synthesis approach shows a great promising technique as a platform for the fabrication of various inorganic/organic hybrid composites.
2. Experimental section
2.1 Materials
Medical absorbent cotton was obtained from Jiaozuo League Hygiene Group (China). All chemicals were analytical grade. Ferrous chloride (FeCl2·4H2O), cobalt chloride hexahydrate (CoCl2·6H2O), sodium hydroxide (NaOH), urea, thiourea, sodium arsenate dibasic (Na2HAsO4·7H2O) and lead nitrate (Pb(NO3)2) were purchased from local chemical agent suppliers. All the solvents and reagents were used without further purification.2.2 Preparation of porous magnetic cellulose beads
The solution with NaOH/thiourea/urea/H2O of 8![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6.5:8:77.5 by weight was cooled to −12 °C. Medical cotton of 2.5 g was immediately added into the above solution under vigorous stirring for 5 min to form a cellulose solution. The solution was then dropwise added into 500 mL freshly prepared aqueous solutions of FeCl2 and CoCl2 with a molar ratio of [Fe]/[Co] = 2 through a 1.4 mm diameter syringe needle. The color of the solution changed immediately from red to black. The beads were kept in the reaction solution for 1 h and then heated to 90 °C for 1 h to promote the formation of iron/cobalt oxyhydroxide complexes. To further stabilize the particles, the beads were filtered and subsequently transferred into 500 mL 1.32 mol L−1 NaOH solution with 5.7 g KNO3 by treating at 90 °C for 3 h.35 The porous magnetic cellulose beads were washed thoroughly with distilled water and ethanol, and then dried by freeze-drying. Porous magnetic cellulose beads prepared from FeCl2 and CoCl2 salts with total concentration of 0.3, 0.5 and 0.7 mol L−1 were labeled as MCB1, MCB2 and MCB3, respectively. Pure cellulose beads prepared from anhydrous ethanol were coded as CB.
:6.5:8:77.5 by weight was cooled to −12 °C. Medical cotton of 2.5 g was immediately added into the above solution under vigorous stirring for 5 min to form a cellulose solution. The solution was then dropwise added into 500 mL freshly prepared aqueous solutions of FeCl2 and CoCl2 with a molar ratio of [Fe]/[Co] = 2 through a 1.4 mm diameter syringe needle. The color of the solution changed immediately from red to black. The beads were kept in the reaction solution for 1 h and then heated to 90 °C for 1 h to promote the formation of iron/cobalt oxyhydroxide complexes. To further stabilize the particles, the beads were filtered and subsequently transferred into 500 mL 1.32 mol L−1 NaOH solution with 5.7 g KNO3 by treating at 90 °C for 3 h.35 The porous magnetic cellulose beads were washed thoroughly with distilled water and ethanol, and then dried by freeze-drying. Porous magnetic cellulose beads prepared from FeCl2 and CoCl2 salts with total concentration of 0.3, 0.5 and 0.7 mol L−1 were labeled as MCB1, MCB2 and MCB3, respectively. Pure cellulose beads prepared from anhydrous ethanol were coded as CB.
2.3 Characterization
A Tensor 27 Fourier transform infrared spectroscopy (FTIR) was used to verify the presence of functional groups in the samples. The morphology of the samples was observed by Hitachi S-4800 scanning electron microscope (SEM) operating at 15 kV. Transmission electron microscopy (TEM) was conducted using JEOL JEM-1011 and high-resolution transmission electron microscopy (HR-TEM) was carried out on JEOL JEM-2011. X-ray photoelectron spectroscopy (XPS) was performed on ESCALab220i-XL electron spectrometer from VG Scientific using 300 W Mg Kα radiation. The nitrogen adsorption–desorption isotherms at 77 K were measured with an automated gas sorption analyzer (Quantachrome Instruments, Autosorb-iQ). The pore size distributions were derived from the desorption branches using the Barrett–Joyner–Halenda (BJH) model. Thermogravimetric analysis (TGA) measurement was performed from room temperature to 600 °C with a heating rate of 10 °C min−1 under steady nitrogen on a PE Pyris. The magnetic property was characterized using a vibrating sample magnetometer (VSM, Lake Shore 7307) at room temperature. Power X-ray diffraction patterns were recorded on a Rigaku D/max 2500 diffractometer with Cu Kα radiation (λ = 1.5406 Å).2.4 Adsorption experiments
Adsorption experiments were carried out in a shaking table at 200 rpm and 25 ± 1 °C using 150 mL shaking flasks containing As(V) or Pb(II) solution with different initial concentrations. The initial concentrations of As(V) and Pb(II) for adsorption isotherm were 5, 10, 20, 30, 40, 50 mg L−1 and 10, 20, 50, 100, 200, 300 mg L−1, respectively. The adsorbent dose was kept as 1 g L−1 for all the experiments. The pH values of all these ions solutions were adjusted to 7.0 ± 0.2 using 0.1 mol L−1 HCl solution. The concentration of ions in the solution was analyzed using an inductively coupled plasma mission spectroscopy (ICPE-9000, Shimadzu). The adsorption capacity qe (mg g−1) was calculated by the following equation.
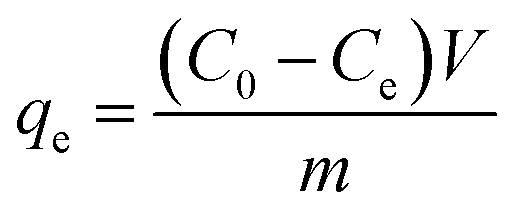 | (1) |
3. Results and discussion
3.1 Formation mechanism of porous magnetic cellulose beads
The porous magnetic cellulose beads were prepared by a facile extrusion of cellulose solution into a coagulation bath of an inorganic solution. The preparation procedure is illustrated in Fig. 1a. Cellulose solution was obtained by dissolving cellulose into NaOH/thiourea/urea aqueous solution, and then added dropwise into iron/cobalt aqueous solution as the coagulation bath. As a result, regenerated cellulose and CoFe2O4 nanoparticles will be formed in situ, resulting in dark magnetic beads. In detail, as shown in Fig. 1b, the coagulation bath can rapidly induce a regenerated cellulose membrane on the surface of the droplet, and the trend will continue into the inner of droplet, leading to the formation of cellulose beads. At the same time, solvent exchange takes place by diffusion from the coagulation bath to the droplet, which can transfer the iron/cobalt ions into the inner of the droplet. Owing to NaOH in the droplet, CoFe2O4 nanoparticles are fabricated by the chemical precipitation reaction of iron/cobalt ions occurring inside the droplet, and thereby tightly entrapped within the porous cellulose beads. In the preparation process, cellulose chains can provide the nuclei-specific crystal growth and serve as nanoreactors for synthesis, further controlling the size and size distribution of generated nanoparticles. Furthermore, cellulose chains stabilize and isolate generated nanoparticles, thus preventing their aggregation.7 The size of as-prepared cellulose beads can be controlled by the syringe needle with different diameter. As shown in Fig. 1c and d, it was seen that the average diameter of MCB is about 3.5 mm, and the size distribution is relatively narrow. Note that the shape of cellulose beads was not perfectly circular. The imperfections probably originate from the high viscous cellulose solution and the strong surface tension between the droplet and the end of the syringe needle. By the way, the as-prepared cellulose beads prepared from anhydrous ethanol collapsed because of a lack of internal support, such as inorganic particles. On the basis of the process above, we can foresee that in addition to iron/cobalt ions, some other metal ions, such as Al(III), Mg(II), Mn(II), Sn(II) and Ce(III), can also be encapsulated into cellulose beads to form macroscopic functional cellulose beads by a facile, cost-effective and safe method. It indicates that the process can be considered to be a universal platform technology to fabricate macroscopic functional cellulose beads.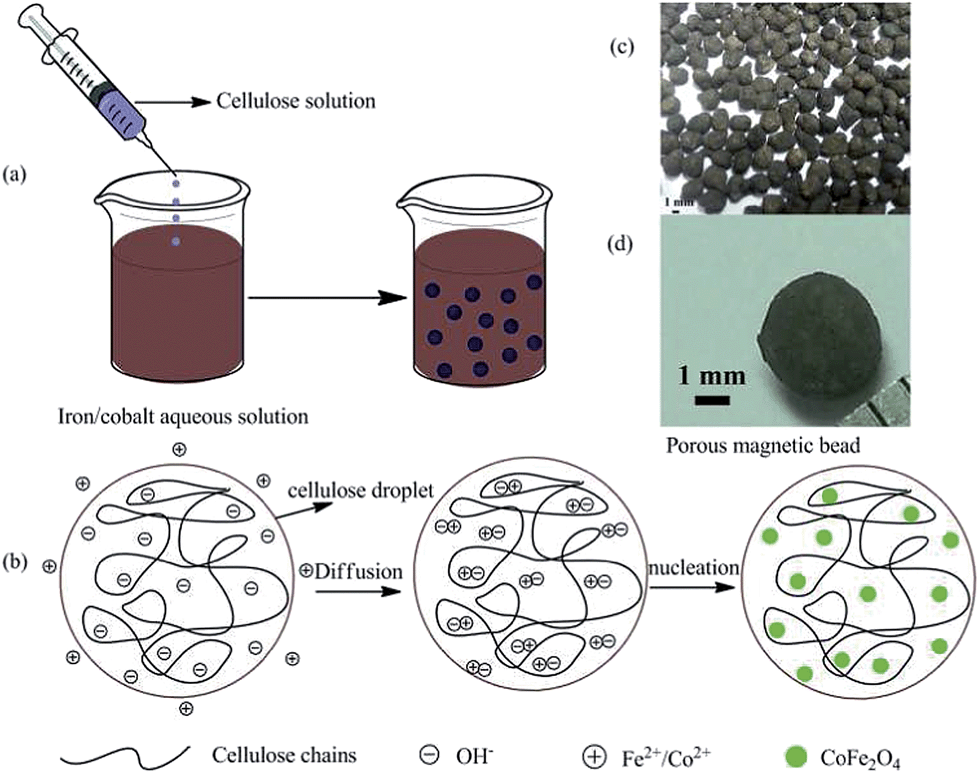 | ||
| Fig. 1 The schematic of the formation procedure of porous magnetic cellulose beads (a) and nucleation of CoFe2O4 nanoparticles (b). Photograph of magnetic cellulose beads (c and d). | ||
3.2 Characterization of porous magnetic cellulose beads
The morphologies of CB and MCB2 are shown in Fig. 2. The surface of cellulose beads exhibits porous structure and high porosity (Fig. 2a and b). As shown in the inset of Fig. 2b, the pore diameter is below 250 nm, and cellulose beads display a sol–gel structure. After the incorporation of inorganic particles, many clustered nanoparticles appear on the surface of MCB2 (Fig. 2c and d), and the inset of Fig. 2d reveals that the sizes of these nanoparticles are ∼50 nm in diameters. SEM images of MCB1 and MCB3 (ESI, Fig. S1†) also show the presence of clustered nanoparticles on the surface. Obviously, these nanoparticles aggregate seriously due to the interparticle dipolar force, and the amount of particles increases with the increase of the concentration of coagulation bath. Surprisingly, some pores were observed on the surface of MCB, indicating that the porous structure of CB is not totally blocked. It is very important for the pollutant diffusion into the inner of beads during the adsorption process. | ||
| Fig. 2 SEM images of CB (a and b) and MCB2 (c and d). Inset is an enlarged image. | ||
To verify the introduction of particles inside cellulose beads, the cross-section SEM images of MCB were performed. As shown in Fig. 3a, SEM image of MCB2 shows that many nanoparticles appear on the surface of cross-section and are dispersed inside cellulose beads throughout the morphology, indicating that iron/cobalt ions have been infused into the inner of cellulose beads through the solvent exchange and cellulose acts as template to prevent CoFe2O4 particles from aggregating. Furthermore, nanoparticles are tightly entrapped within cellulose beads (indicated by the red circle in Fig. 3b) owing to the interaction between cellulose and inorganic particles. Many pores were observed on the surface of cross-section in Fig. 3c, revealing that there are numerous interpenetrating pores inside MCB2. Similar results were observed from cross-section SEM images of MCB1 (ESI, Fig. S2†). However, particles in MCB3 display strong aggregation compared with MCB1 and MCB2, which can be attributed to the high amount of iron and cobalt in coagulation bath. Thus, in combination with these inorganic nanoparticles and pores, cellulose beads can not only obtain the good mechanical property, but facilitate the transport of pollutant from the solution to the inner of MCB. From the corresponding size distribution histogram (Fig. 3d; ESI, Fig. S2d and S3d†), the average particles size of CoFe2O4 for MCB1, MCB2 and MCB3 is 200, 290, and 210 nm, respectively, which is larger than that of pure CoFe2O4.36 It is due to the fact that cellulose chain can act as nuclei, thus the growth process is spontaneous and fast, resulting in the larger particles.
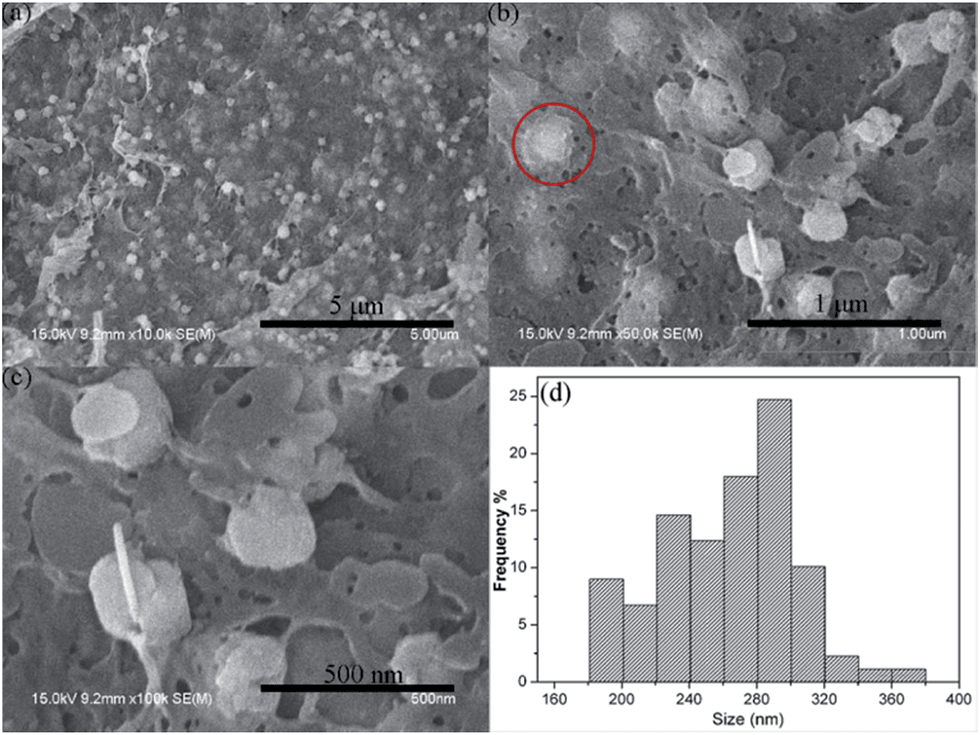 | ||
| Fig. 3 Cross-section SEM images of MCB2 (a, b and c) and size distribution of CoFe2O4 nanoparticles (d). | ||
To observe the nanoparticles more clearly by TEM, cellulose template was removed by calcination at 300 °C. Low-magnification TEM images reveal that many particles with cubical shapes were observed in the TEM images of MCB (Fig. 4a; ESI, Fig. S4a and c†). Interestingly, some smaller particles with a size of around 25 nm were present in the sample MCB, as typically indicated by the red circle for MCB2 in Fig. 4a, which should originate from the surface of MCB2. It is consistent with the observation of surface SEM. From the high-magnification TEM images of MCB (Fig. 4b; ESI, Fig. S4b and d†), the particle sizes of MCB1, MCB2 and MCB3 are all in the range of 200–300 nm. Surprisingly, many void space and mesoporous structures throughout the particles can be observed in the TEM image of MCB2 (Fig. 4b), confirming that the large particles are the assemblies of small particles.37 The high-resolution TEM (HR-TEM) of a single particle from MCB2 (Fig. 4c) shows the visible lattice fringes with an interplanar distance of 0.475 nm, corresponding to the (111) reflections of the spinel phase.35,36 The selected area electron diffraction (SAED) pattern (Fig. 4d) shows diffuse rings that can be indexed to standard CoFe2O4 diffraction data.
 | ||
| Fig. 4 Low-magnification TEM image (a), high-magnification TEM image (b), HR-TEM image (c) and SAED pattern of MCB2 (d). | ||
To further clear out the pore structure of samples, the nitrogen adsorption–desorption isotherms were performed. As shown in Fig. 5, the isotherms of all samples exhibit typical type III isotherm with distinct hysteresis loops close to H3 type, indicating that there may be mesoporous structure,38,39 which has been seen more clearly in the high-magnification TEM image in Fig. 4b. The specific surface area, pore volume, and average pore size of these samples are listed in Table 1. Compared with these samples, a notable reduction of the specific surface areas, the pore volumes and average pore size was observed. It is due to the formation of CoFe2O4 nanoparticles on the surface and inner of CB, leading to the block of some pore structures. Furthermore, it was observed that the specific surface area of MCB increases with the increase of the content of CoFe2O4 nanoparticles, which can be attributed to the high amount of incorporated CoFe2O4 nanoparticles that has the large specific surface area. Interestingly, it was seen that MCB3 presented the high pore volume and contained broadly distributed pores with small mesopores (3.140 nm) and larger ones (15.250 nm). The larger pores may be due to the void space of surface nanoparticles. The nitrogen adsorption–desorption results suggest that there is a good pore structure in the MCB and the pore structure can be reserved after incorporating CoFe2O4 nanoparticles.
 | ||
| Fig. 5 Nitrogen adsorption–desorption isotherm of CB (a), MCB1 (b), MCB2 (c) and MCB3 (d); inset is the BJH pore size distribution. | ||
| Sample | SBET (m2 g−1) | Vt (cm3 g−1) | D (nm) |
|---|---|---|---|
| CB | 68.520 | 0.486 | 19.665 |
| MCB1 | 42.302 | 0.163 | 3.934 |
| MCB2 | 47.812 | 0.161 | 3.935 |
| MCB3 | 57.712 | 0.205 | 3.140, 15.250 |
XRD was used to identify the phase structure of samples. As shown in Fig. 6a, CB presented typical cellulose II structure with two diffraction peaks at 20.1° and 21.7°. For MCB, besides the typical peaks of cellulose II structure, there are some other diffraction peaks at 2θ = 18.3°, 30.2°, 35.6°, 37.1°, 43.2°, 53.4°, 57.0°, 62.5°, 70.9°, 74.0°, 75.0° and 78.8°, corresponding to the (111), (220), (311), (222), (400), (422), (511), (440), (620), (533), (622) and (551) planes of CoFe2O4 with the spinel phase (JCPDS card no. 22-1086), respectively. Note that an oxyhydroxide phase with a structure similar to α-FeOOH (JCPDS card no. 29-713) was observed from XRD cure of MCB3. This may be due to hydrolysis of excess ferrous ion to form α-FeOOH on the surface of cellulose beads. According to the Scherrer equation, the crystallite sizes of CoFe2O4 nanoparticles is ∼19, 23 and 25 nm for MCB1, MCB2 and MCB3, respectively. These are in agreement with the particle sizes obtained from the surface SEM. The XRD results indicate that magnetic cellulose beads are composed of cellulose and ferromagnetic cobalt ferrite nanoparticles.
 | ||
| Fig. 6 XRD curves (a), FTIR spectra (b) and TG curves (c) of CB, MCB1, MCB2 and MCB3; the magnetic hysteresis cures of MCB1, MCB2 and MCB3 (d). Inset: dispersion and magnetic separation of MCB in distilled water. | ||
FTIR spectra of CB, MCB1, MCB2 and MCB3 were presented in Fig. 6b. The spectra of samples display the typical characteristic peaks of cellulose II.40 The absorption peak at 3415 cm−1 is assigned to OH stretching vibration of cellulose II. The band at 2892 cm−1 corresponds to CH2 symmetrical stretching band and CH2 symmetrical bending band was observed at 1420 cm−1. The band at 1160 cm−1 is due to antisymmetrical bridge C–O–C stretching mode, and the band attributed to the skeletal stretching vibrations of C–O bond appears at 1066 cm−1. The band at 896 cm−1 relates to the β-glycosidic linkages in cellulose. The absorption peaks at 1374, 1338, 1318, and 1265 cm−1 are due to C–H bending, O–H in-plane bending, CH2 wagging and C–H bending, respectively. Two new bands in the spectra of MCB appear at 585 and 420 cm−1 that can be assigned to Fe(Co)–O characteristic peaks of CoFe2O4, corresponding to the intrinsic lattice vibrations of octahedral and tetrahedral coordination compounds in the spinel structure, respectively.41 The differences in the vibration frequency are probably due to the long Fe(Co)–O distances originating from the octahedral sites and the short Fe(Co)–O distances in the tetrahedral sites.41,42
TG curves of CB and MCB were shown in Fig. 6c. It is apparent that the curve of CB displays a large stage of weight loss that is due to the decomposition of cellulose, and the residual weight is 2.36%. The curves of MCB also exhibit a stage of weight loss, but the weight loss is small compared with the weight loss of cellulose. The difference can be attributed to the different amount of CoFe2O4 in the samples. According to TG cures, the residual weight for MCB1, MCB2 and MCB3 is 22.68%, 28% and 43.15%, respectively. The total weight fraction of CoFe2O4 can further be estimated as 20.32%, 25.64% and 40.79% for MCB1, MCB2 and MCB3, respectively.
The room-temperature hysteresis measurements of MCB were carried out in the applied magnetic field sweeping from −10 to 10 kOe. As shown in Fig. 6d, the magnetic samples MCB exhibit the low saturation magnetization MS and large coercivities HC, which is probably due to the prominent growth of magnetic anisotropy inhibiting the alignment of the moment in an applied field.43 The detailed values of the saturation magnetization (MS), remanence (MR) and coercivities (HC) are summarized in Table 2. It is seen that the values of MS are much lower than the bulk values of CoFe2O4 (80.8 emu g−1),43 which can be explained by cation inversion, vacancies and non-stoichiometric composition typical of water-based synthesis of CoFe2O4 nanoparticles from (oxy)-hydroxides.35 However, HC values of samples are in agreement with the values of similar size of CoFe2O4 nanoparticles, and increase as the particle size increases. The small MR reveals a large number of reversible processes,44 and the squareness ratios (MR/MS) in the range 0.438–0.517 are very close to that expected value for the cobalt ferrite nanoparticles of non-interacting single domain with uniaxial anisotropy.43 The inset shows that the magnetic MCB can possess a sensitive magnetic response with external magnetic field, thus it can be easily separated from aqueous solution in some application.
| Sample | DXRD (nm) | MS (emu g−1) | MR (emu g−1) | HC (Oe) | MR/MS |
|---|---|---|---|---|---|
| MCB1 | 19 | 13.57 | 5.94 | 935 | 0.438 |
| MCB2 | 23 | 16.06 | 7.79 | 992 | 0.485 |
| MCB3 | 25 | 16.70 | 8.63 | 1111 | 0.517 |
XPS analysis was used to investigate the surface components of samples. In the wide spectra (ESI, Fig. S5a†), Co 2p and Fe 2p signals in the MCB XPS spectra clearly appear at about 780 and 710 eV, respectively, in addition to C 1s and O 1s signals (located at about 284.8 and 532 eV, respectively) compared with that of CB XPS spectrum. As shown in Fig. 7a, the C 1s spectrum of MSB2 exhibited three peaks at 284.8, 286.4 and 287.7 eV, arising from C–C, C–O and O–C–O of cellulose, respectively, which is in agreement with the XPS spectra of CB, MSB1 and MSB3 in Fig. S5b–d (ESI).† The O 1s spectrum of CB (ESI, Fig. S5e†) can be deconvoluted into two peaks located at 532.1 and 533.0 eV, which can be attributed to oxygen atoms in the cellulose (C–O) and in the adsorbed water (–OH),45 respectively. However, the O 1s spectrum of MSB2 (Fig. 7b) appears a new peak of Fe/Co–O with binding energy of 530.6 eV besides the above-mentioned peaks. In the spectrum of Co 2p (Fig. 7c), the Co 2p peaks can be assigned to Co 2p3/2 and its satellites located at 781.2 and 786.7 eV, and Co 2p1/2 and its satellites located at 797.1 and 803.1 eV, respectively. The Fe 2p spectrum of MSB2 exhibits two peaks with binding energies of 711.6 and 725.1 eV, as shown in Fig. 7d, which can be attributed to Fe 2p3/2 and Fe 2p1/2, respectively. Similarly, the analysis of MSB1 and MSB2 (ESI, Fig. S5f–i†) also show the same results with MSB2, thus confirming the presence of CoFe2O4 in the cellulose matrix.
 | ||
| Fig. 7 C 1s (a), O 1s (b), Co 2p (c) and Fe 2p (d) XPS spectra of MCB2. | ||
3.3 Adsorption experiment
Hence, inspired by the advantage of high specific surface area, robust interconnected networks and easy separation from aqueous solution, such a porous material can act as a promising candidate for the removal of pollutants. Langmuir and Freundlich adsorption isotherms are used to estimate the adsorption process. Langmuir isotherm is expressed as follows:
 | (2) |
Freundlich isotherm is represented by the following equation:
| qe = KfCe1/n | (3) |
Langmuir and Freundlich adsorption isotherms of As(V) and Pb(II) were shown in Fig. 8, and the adsorption parameters were listed in Table S1 (ESI).† The high correlation coefficients (R2 > 0.97) show that Langmuir isotherm can describe the adsorption process well, revealing that the adsorption is a monolayer adsorption process. MCB3 displays the maximal adsorption capacities for As(V) and Pb(II), 20.27 and 97.29 mg g−1, respectively, indicating that CoFe2O4 particles should be the effective component to adsorb metal ions. These values are much higher than those of magnetite-reduced graphene oxide composites14 (5.83 mg g−1 for As(V)), mesoporous magnetic iron oxide@carbon encapsulates21 (17.9 mg g−1 for As(V)), urchin-like α-FeOOH hollow spheres39 (80 mg g−1 for Pb(II)), magnetic porous ferrospinel MnFe2O4 (69 mg g−1 for Pb(II))46 and oxidized multiwalled carbon nanotubes20 (54.11 mg g−1 for Pb(II)). In addition, most of above-mentioned adsorption capacities were obtained at optimal pH values that had a significant effect on the adsorption for metal ions. Thus, it is more reasonable to estimate the potential capability of our prepared adsorbent for metal ions removal in the practical water treatment, because the adsorption was performed at pH 7 that agrees with pH of practical water. In addition, Freundlich constant n is greater than 1, which is a favorable condition for adsorption.
 | ||
| Fig. 8 Langmuir (a and c) and Freundlich (b and d) isotherms of As(V) (a and b) and Pb(II) (c and d) on the MCB (pH = 7). | ||
The adsorption kinetics describing the metal ions uptake rate is used to understand the adsorption property of metal ions on MCB. As shown in Fig. S6a and b (ESI),† the adsorption rates of As(V) and Pb(II) are relatively fast within 60 min, and then slow down until the adsorption reaches equilibrium after 360 min. The rapid adsorption rate may be due to the larger pores in MCB, which result in the rapid transport of metal ions into the adsorption sites inside MCB. The pseudo-second-order and intra-particle diffusion kinetic model were employed to fit the experimental data, respectively, which can be expressed as:
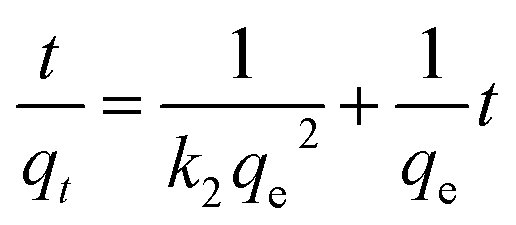 | (4) |
| qt = kidt1/2 + C | (5) |
4. Conclusions
In summary, we have developed a facile one-pot, “green” method for the fabrication of the functional macroscopic cellulose beads via simple wet extrusion. The functional cellulose beads exhibit abundant pore structure and sensitive magnetic induced behavior. CoFe2O4 particles are tightly entrapped into the pores of cellulose beads. The entrapped active particles can increase the adsorption capacities of As(V) and Pb(II) onto cellulose beads. Considering the versatility of wet extrusion, we foresee that the entrapped active components can be extended to a multitude of other functional substrates, such as iron oxide, magnesium hydroxide, aluminium hydroxide, even dyes, medicine and enzyme, for different applications.Acknowledgements
This project was supported by the National Basic Research Program of China (973 Program, no. 2011CB933700) of Ministry of Science and Technology of China.Notes and references
- R. B. Thompson, V. V. Ginzburg, M. W. Matsen and A. C. Balazs, Science, 2001, 292, 2469–2472 CrossRef CAS PubMed.
- M. S. Hu, H. L. Chen, C. H. Shen, L. S. Hong, B. R. Huang, K. H. Chen and L. C. Chen, Nat. Mater., 2006, 5, 102–106 CrossRef CAS PubMed.
- D. X. Li, Y. Cui, K. W. Wang, Q. He, X. H. Yan and J. B. Li, Adv. Funct. Mater., 2007, 17, 3134–3140 CrossRef CAS.
- J. Virkutyte and R. S. Varma, Chem. Sci., 2011, 2, 837–846 RSC.
- J. B. Lian, X. C. Duan, J. M. Ma, P. Peng, T. I. Kim and W. J. Zheng, ACS Nano, 2009, 3, 3749–3761 CrossRef CAS PubMed.
- A. Bakandritsos, A. B. Bourlinos, V. Tzitzios, N. Boukos, E. Devlin, T. Steriotis, V. Kouvelos and D. Petridis, Adv. Funct. Mater., 2007, 17, 1409–1416 CrossRef CAS.
- B. A. Rozenberg and R. Tenne, Prog. Polym. Sci., 2008, 33, 40–112 CAS.
- W. Z. Yuan, J. Y. Yuan, F. B. Zhang and X. M. Xie, Biomacromolecules, 2007, 8, 1101–1108 CrossRef CAS PubMed.
- Z. Liu, H. S. Wang, B. Li, C. Liu, Y. J. Jiang, G. Yu and X. D. Mu, J. Mater. Chem., 2012, 22, 15085–15091 RSC.
- X. G. Luo and L. N. Zhang, Biomacromolecules, 2010, 11, 2896–2903 CrossRef CAS PubMed.
- X. L. Yu, S. R. Tong, M. F. Ge, J. C. Zuo, C. Y. Cao and W. G. Song, J. Mater. Chem. A, 2013, 1, 959–965 CAS.
- F. Mou, J. Guan, H. Ma, L. Xu and W. Shi, ACS Appl. Mater. Interfaces, 2012, 4, 3987–3993 CAS.
- J. S. Hu, L. S. Zhong, W. G. Song and L. J. Wan, Adv. Mater., 2008, 20, 2977–2982 CrossRef CAS.
- V. Chandra, J. Park, Y. Chun, J. W. Lee, I. C. Hwang and K. S. Kim, ACS Nano, 2010, 4, 3979–3986 CrossRef CAS PubMed.
- C. Gao, X. Y. Yu, R. X. Xu, J. H. Liu and X. J. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 4672–4682 CAS.
- X. Y. Yu, R. X. Xu, C. Gao, T. Luo, Y. Jia, J. H. Liu and X. J. Huang, ACS Appl. Mater. Interfaces, 2012, 4, 1954–1962 CAS.
- C.-Y. Cao, J. Qu, F. Wei, H. Liu and W.-G. Song, ACS Appl. Mater. Interfaces, 2012, 4, 4283–4287 CAS.
- L. H. Ai, H. T. Yue and J. Jiang, Nanoscale, 2012, 4, 5401–5408 RSC.
- X. Y. Yu, T. Luo, Y. Jia, Y. X. Zhang, J. H. Liu and X. J. Huang, J. Phys. Chem. C, 2011, 115, 22242–22250 CAS.
- X. Y. Yu, T. Luo, Y. X. Zhang, Y. Jia, B. J. Zhu, X. C. Fu, J. H. Liu and X. J. Huang, ACS Appl. Mater. Interfaces, 2011, 3, 2585–2593 CAS.
- Z. X. Wu, W. Li, P. A. Webley and D. Y. Zhao, Adv. Mater., 2012, 24, 485–491 CrossRef CAS PubMed.
- X. L. Yu, S. R. Tong, M. F. Ge and J. C. Zuo, Carbohydr. Polym., 2013, 92, 269–275 CrossRef CAS PubMed.
- S. D. Jiang, Q. Z. Yao, G. T. Zhou and S. Q. Fu, J. Phys. Chem. C, 2012, 116, 4484–4492 CAS.
- X. L. Yu, S. R. Tong, M. F. Ge, L. Y. Wu, J. C. Zuo, C. Y. Cao and W. G. Song, Carbohydr. Polym., 2013, 92, 380–387 CrossRef CAS PubMed.
- Y. Tian, M. Wu, R. G. Liu, Y. X. Li, D. Q. Wang, J. J. Tan, R. C. Wu and Y. Huang, Carbohydr. Polym., 2011, 83, 743–748 CrossRef CAS PubMed.
- I. F. Nata, M. Sureshkumar and C. K. Lee, RSC Adv., 2011, 1, 625–631 RSC.
- X. L. Cheng, J. S. Jiang, M. Hu, G. Y. Mao, Z. W. Liu, Y. Zeng and Q. H. Zhang, CrystEngComm, 2012, 14, 3056–3062 RSC.
- L. H. Han, Y. C. Chen and Y. Wei, CrystEngComm, 2012, 14, 4692–4698 RSC.
- J. B. Fei, Y. Cui, J. Zhao, L. Gao, Y. Yang and J. B. Li, J. Mater. Chem., 2011, 21, 11742–11746 RSC.
- C. C. Yu, X. P. Dong, L. M. Guo, J. T. Li, F. Qin, L. X. Zhang, J. L. Shi and D. S. Yan, J. Phys. Chem. C, 2008, 112, 13378–13382 CAS.
- C. Y. Cao, J. Qu, W. S. Yan, J. F. Zhu, Z. Y. Wu and W. G. Song, Langmuir, 2012, 28, 4573–4579 CrossRef CAS PubMed.
- B. Fadeel and A. E. Garcia-Bennett, Adv. Drug Delivery Rev., 2010, 62, 362–374 CrossRef CAS PubMed.
- A. Elsaesser and C. V. Howard, Adv. Drug Delivery Rev., 2012, 64, 129–137 CrossRef CAS PubMed.
- T. R. Pisanic, J. D. Blackwell, V. I. Shubayev, R. R. Finones and S. Jin, Biomaterials, 2007, 28, 2572–2581 CrossRef CAS PubMed.
- R. T. Olsson, M. Samir, G. Salazar-Alvarez, L. Belova, V. Strom, L. A. Berglund, O. Ikkala, J. Nogues and U. W. Gedde, Nat. Nanotechnol., 2010, 5, 584–588 CrossRef CAS PubMed.
- G. Salazar-Alvarez, R. T. Olsson, J. Sort, W. A. A. Macedo, J. D. Ardisson, M. D. Baro, U. W. Gedde and J. Nogues, Chem. Mater., 2007, 19, 4957–4963 CrossRef CAS.
- H. L. Yuan, Y. Q. Wang, S. M. Zhou, L. S. Liu, X. L. Chen, S. Y. Lou, R. J. Yuan, Y. M. Hao and N. Li, Nanoscale Res. Lett., 2010, 5, 1817–1821 CrossRef CAS PubMed.
- M. Kruk and M. Jaroniec, Chem. Mater., 2001, 13, 3169–3183 CrossRef CAS.
- B. Wang, H. B. Wu, L. Yu, R. Xu, T. T. Lim and X. W. Lou, Adv. Mater., 2012, 24, 1111–1116 CrossRef CAS PubMed.
- M. L. Nelson and R. T. O'Connor, J. Appl. Polym. Sci., 1964, 8, 1311–1324 CrossRef CAS.
- A. Pui, D. Gherca and G. Carja, Dig. J. Nanomater. Biostruct., 2011, 6, 1783–1791 Search PubMed.
- P. Tailhades, C. Villette, A. Rousset, G. U. Kulkarni, K. R. Kannan, C. N. R. Rao and M. Lenglet, J. Solid State Chem., 1998, 141, 56–63 CrossRef CAS.
- K. Maaz, A. Mumtaz, S. K. Hasanain and A. Ceylan, J. Magn. Magn. Mater., 2007, 308, 289–295 CrossRef CAS PubMed.
- M. P. Gonzalez-Sandoval, A. M. Beesley, M. Miki-Yoshida, L. Fuentes-Cobas and J. A. Matutes-Aquino, J. Alloys Compd., 2004, 369, 190–194 CrossRef CAS PubMed.
- L. S. Johansson, J. M. Campbell, K. Koljonen and P. Stenius, Appl. Surf. Sci., 1999, 144–45, 92–95 CrossRef.
- Y. M. Ren, N. Li, J. Feng, T. Z. Luan, Q. Wen, Z. S. Li and M. L. Zhang, J. Colloid Interface Sci., 2012, 367, 415–421 CrossRef CAS PubMed.
- R. P. Han, L. J. Zhang, C. Song, M. M. Zhang and H. M. Zhu, Carbohydr. Polym., 2010, 79, 1140–1149 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05601a |
| This journal is © The Royal Society of Chemistry 2014 |