
Unexpected hydrazine hydrate-mediated aerobic oxidation of aryl/ heteroaryl boronic acids to phenols in ambient air†
Yanzhen Zhong,
Linxin Yuan,
Zheng Huang,
Wenchao Gu,
Ye Shao and
Wei Han*
Jiangsu Collaborative Innovation Center of Biomedical Functional Materials, Jiangsu Key Laboratory of Biofunctional Materials, Key Laboratory of Applied Photochemistry, School of Chemistry and Materials Science, Nanjing Normal University, Wenyuan Road NO.1, 210023 Nanjing, China. E-mail: whhanwei@gmail.com; Fax: +86-25-8589-1455
First published on 16th July 2014
Abstract
The expedient and efficient sub-stoichimetric hydrazine hydrate-mediated aerobic hydroxylation of boronic acids that proceeds in poly(ethylene glycol) (PEG-400) has been successfully developed, providing diverse phenols in high yields. And heteroaryl boronic acids are also amenable to this protocol.
The phenol motif is widespread in a vast array of polymers, pharmaceuticals, and naturally occurring compounds.1 Moreover, it often serves as the key building block for construction of more complex structures. As a result, establishing practical, general, and efficient catalytic methods for the synthesis of phenols remain an area of tremendous research efforts.
Among the methods developed to prepare phenols, the oxidative ipso-hydroxylation of arylboronic acids has attracted considerable interest because arylboronic acids are readily available, thermally-, air- and water-stable, and easily separated from products. Furthermore, classical phenol synthesis methods, i.e. nucleophilic aromatic substitution of aryl halides, hydrolysis of arene diazonium salts, and benzyne protocols,2 frequently suffer from low functional group compatibility, poor accessibility of the starting materials, and harsh reaction conditions. Transition-metal-catalysis as a powerful tool has been widely applied in organic synthesis and also introduced to oxidative hydroxylation of arylboronic acids. For instance, a variety of copper, ruthenium, or palladium catalysts have been developed for this transformation and achieved effective catalysis for a broad substrate scope (Scheme 1a)3 under air or oxygen atmosphere. From a practical point of view, it is highly desirable to develop nonmetal-catalyzed processes in the pharmaceutical industry, since metal contamination can cause severe concerns and metal removal can be cumbersome. Although numerous nonmetal-mediated processes have been utilized in hydroxylation of arylboronic acids, most of them require stoichiometric strong oxidants, as shown in the Scheme 1b4–13 and often lead to poor functional group tolerance, safety issues and/or stoichiometric amounts of unwanted byproducts. Compared to these oxidants, air and molecular oxygen are the ideal oxidants with low cost and lack of toxic byproducts.14
 | ||
| Scheme 1 Oxidative ipso-hydroxylation of boronic acids. | ||
During the preparation of this manuscript, Xiao and co-workers reported a stoichiometric methylhydrazine-mediated hydroxylation of arylboronic acids using air as oxidant (Scheme 1c).15 And subsequently, a complicated organic catalyst was reported to catalyze this transformation with the assistant of stoichiometric hydrazine hydrate in the presence of molecular oxygen (Scheme 1d).16 And, in the both methods toxic and volatile solvents were used as reaction media. Herein, we disclose the first inexpensive sub-stoichimetric hydrazine hydrate-mediated oxidative ipso-hydroxylation of arylboronic acids to phenols by using ambient air as the sole oxidant in green solvent poly(ethylene glycol) (Scheme 1e). A remarkable feature of this protocol is the compatibility of heteroaryl boronic acids which remains elusive.12
Recently, we have smoothly launched ligand-free catalytic systems for cross-coupling reactions,17 utilizing PEG-400 [poly(ethylene glycol) with an average molecular weight of 400 Da]18 as a solvent. This report is based on unexpected findings during the exploration of carbon–nitrogen bond-forming reactions of hydrazine hydrate with arylboronic acids in PEG-400. During the exploration, we observed hydrazine hydrate promoted ipso-hydroxylation of arylboronic acids to phenols in the absence of a transition-metal catalyst under air atmosphere. Encouraged by this observation, we first investigated hydrazine hydrate-promoted aerobic oxidation of dibenzothiophen-4-ylboronic acid (1a) to dibenzothiophen-4-ol (2a) in the absence of a metal catalyst. The structure of 2a was proved by single-crystal X-ray diffraction.19 Selected results from our screening experiments are presented in Table 1. The bases affected the reaction. For example, K3PO4, and K2CO3 gave moderate results (Table 1, entries 1 and 5), whereas Na2CO3, KOH, NaF, TBAF, and Et3N were found to be ineffective (Table 1, entries 4, 6, 7, 8, and 9).
|
||||
|---|---|---|---|---|
| Entry | [cat] (mol %) | Base | Water (equiv.) | Yieldb of 2a/% |
| a Reaction conditions (unless otherwise stated): 1a (0.5 mmol), base (1.0 mmol), PEG-400 (2.0 g), air, 80 °C, 6 h.b Isolated yield after flash chromatography.c 24 h.d O2 (balloon). | ||||
| 1 | N2H4·H2O (200) | K3PO4 | — | 54 |
| 2 | N2H4·H2O (200) | Cs2CO3 | — | 79 |
| 3 | N2H4·H2O (200) | CH3COOK | — | 70 |
| 4 | N2H4·H2O (200) | Na2CO3 | — | <5 |
| 5 | N2H4·H2O (200) | K2CO3 | — | 52 |
| 6 | N2H4·H2O (200) | KOH | — | Trace |
| 7 | N2H4·H2O (200) | NaF | — | 38 |
| 8 | N2H4·H2O (200) | TBAF | — | <5 |
| 9 | N2H4·H2O (200) | Et3N | — | Trace |
| 10 | N2H4·H2O (200) | Cs2CO3 | H2O (1) | 83 |
| 11 | N2H4·H2O (200) | Cs2CO3 | H2O (3) | 87 |
| 12 | N2H4·H2O (200) | Cs2CO3 | H2O (5) | 91 |
| 13 | N2H4·H2O (200) | Cs2CO3 | H2O (10) | 78 |
| 14 | — | Cs2CO3 | H2O (5) | 29 |
| 15 | N2H4·H2O (50) | Cs2CO3 | H2O (5) | 43 |
| 16c | N2H4·H2O (50) | Cs2CO3 | H2O (5) | 86 |
| 17c | N2H4·H2O (20) | Cs2CO3 | H2O (5) | 83 |
| 18c | N2H4·H2O (50) | — | H2O (5) | 41 |
| 19d | N2H4·H2O (50) | Cs2CO3 | H2O (5) | 96 |
| 20c | C6H5NHNH2 (50) | Cs2CO3 | H2O (5) | 10 |
| 21c | C6H5NH2 (50) | Cs2CO3 | H2O (5) | 10 |
| 22c | NH3·H2O (50) | Cs2CO3 | H2O (5) | Trace |

Comparatively, Cs2CO3 gave a better result (Table 1, entry 2). The effect of water on the reaction was also evaluated. The best result was obtained when 5.0 equiv. of water was used (Table 1, entry 12). However, in the absence of hydrazine hydrate, the yield was decreased to 29% (Table 1, entry 14). Gratifyingly, a sub-stoichimetric amount of hydrazine hydrate (50 mol%) and (20 mol%) also gave a satisfactory yield (86%) and (83%) in 24 h, respectively (Table 1, entries 16 and 17). Under otherwise equal reaction conditions this transformation was performed under an oxygen atmosphere and afforded 2a in 96% yield in 6 h (Table 1, entry 18). Considering operability and practicability, we chose air as oxidant. Other catalysts such as phenylhydrazine, aniline, and NH3·H2O were ineffective with this reaction (Table 1, entries 20–22).
Having identified the optimal conditions, we next explored the efficacy of hydrazine hydrate-mediated aerobic oxidation of various arylboronic acids to phenols (Scheme 2). A wide array of arylboronic acids could be oxidized to phenols in good to excellent yields. The electronic properties of the arylboronic acids was found to be only loosely correlated with their reactivity and had little effect on this transformation. Sterically hindered arylboronic acids (e.g., 2b, 2d, 2e, and 2i) were well-tolerated. Base-sensitive functional groups methyl ketone and ester also proceeded successfully, leading to desire products 2o and 2p in 95% and 97% yields, respectively. Notably, an oxidation-sensitive free aniline group proved to be a compatible functional group (e.g., 2h), which was rarely reported in this type of transformation.20 And, the product 3-aminophenol (2h) is a key intermediate for the synthesis of fluorescent dye Rhodamine B. Furthermore, iodo and bromo substitutes that have been widely applied in cross-coupling reactions were well tolerated with this method (e.g., 2m and 2n). To our delight, not only boronic acids but a boronic ester smoothly converted to corresponding phenol 2p in 97% yield. We were also pleased to find that 2-naphthylboronic acid underwent an efficient aerobic oxidative ipso-hydroxylation (e.g., 2s). Significantly, sulfide 2t, an oxidation-sensitive substrate, can tolerate the normal conditions without suffering overoxidation.
 | ||
| Scheme 2 Hydrazine hydrate-mediated aerobic oxidation of arylboronic acid. Reaction conditions (unless otherwise stated): 1 (0.5 mmol), Cs2CO3 (1.0 mmol), H2O (2.5 mmol), N2H4·H2O (0.25 mmol), PEG-400 (2.0 g), 80 °C, air. Isolated yield after flash chromatography. [a] With 4-carbomethoxy_phenylboronic acid pinacol ester. | ||
Notably, heteroarylboronic acids are also amenable to this protocol. Various heteroaryl systems were explored, including dibenzothiophenyl, dibenzofuranyl, thiophenyl, pyridinyl, and indazolyl derivatives. And, the corresponding heteroarylboronic acids delivered desired products with superior yields (Table 2).
| Entry | Boronic acid | Product | Time/h | Yield/% |
|---|---|---|---|---|
| a Reaction conditions (unless otherwise stated): 1 (0.5 mmol), Cs2CO3 (1.0 mmol), H2O (2.5 mmol), N2H4·H2O (0.25 mmol), PEG-400 (2.0 g), 80 °C, air. Isolated yield after flash chromatography. | ||||
| 1 |  |
 |
24 | 86 |
| 2 |  |
 |
12 | 83 |
| 3 |  |
 |
24 | 91 |
| 4 |  |
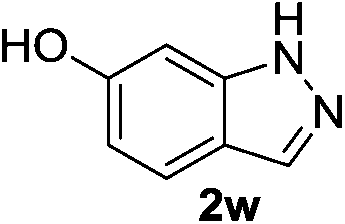 |
12 | 96 |
To gain insight into the possible mechanism, control experiments were carried out. When the model reaction was performed in anhydrous conditions under oxygen atmosphere, a excellent yield of 2a can be obtained [eqn (1)]. Whereas the same reaction proceeded in oxygen-free system, 2a can not be detected [eqn (2)]. Furthermore, under normal conditions H2O was replaced by H218O and as a result, the product was still the 2a [eqn (3)]. These results suggest that molecular oxygen rather than water is the donor of the oxygen atom in the oxidative hydroxylation reactions. Recently, Berkessel et al. reported multiple H-bond dramatically accelerating olefin epoxidation.21 On the basis of above results and previous studies,4b,21,22 the reaction presumably involves hydrazine hydrate firstly activating oxygen molecule through hydrogen bonding. Subsequently the activated oxygen molecule undergoes nucleophilic attack on boronic acid I to form a key intermediate II (or III). Finally, phenyl migration and hydrolysis provide the final product (Scheme 3).
 | ||
| Scheme 3 Plausible mechanism for hydrazine hydrate-mediated oxidative ipso-hydroxylation of aryl/heteroaryl boronic acids. | ||
In summary, a general, practical and efficient hydrazine hydrate-mediated hydroxylation of boronic acids and a boronate ester has been developed.23 The use of air as the sole oxidant makes the process both safely and environmentally sound. Substrates bearing electron-withdrawing or electron-donating functionality, active groups, as well as ortho-substitution underwent smoothly, delivering phenols in high yields. Additionally, heteroaryl boronic acids are also well tolerated in this protocol.
The work was sponsored by the Natural Science Foundation of China (21302099), the Natural Science Foundation of Jiangsu Province (BK2012449), the Natural Science Foundation of Jiangsu Provincial Colleges and Universities (12KJB150014), the Scientific Research Start-up Foundation of Nanjing Normal University (2011103XGQ0250), the SRF for ROCS, SEM, and the Priority Academic Program Development of Jiangsu Higher Education Institutions. We thank Prof. Yaqian Lan and Dr Shunli Li for assistance with X-ray structure analysis.
Notes and references
- (a) J. H. P. Tyman, Synthetic and Natural Phenols, Elsevier, New York, 1996 Search PubMed; (b) Z. Rappoport, The Chemistry of Phenols, Wiley-VCH, Weinheim, 2003 CrossRef.
- (a) K. G. Thakur and G. Sekar, Chem. Commun., 2011, 47, 6692 RSC; (b) A. Mehmood and N. E. Leadbeater, Catal. Commun., 2010, 12, 64 CrossRef CAS PubMed; (c) C. Hoarau and T. R. R. Pettus, Synlett, 2003, 127 CAS; (d) P. Hanson, J. R. Jones, A. B. Taylor, P. H. Walton and A. W. Timms, J. Chem. Soc., Perkin Trans. 2, 2002, 1135 RSC; (e) T. George, R. Mabon, G. Sweeney, B. J. Sweeney and A. Tavassoli, J. Chem. Soc., Perkin Trans. 1, 2000, 2529 RSC; (f) C. A. Fyfe, The Chemistry of the Hydroxyl Group, ed. S. Patai, Wiley Interscience, New York, 1971, vol. 1, p. 83 Search PubMed.
- (a) Y.-Q. Zou, J.-R. Chen, X.-P. Liu, L.-Q. Lu, R. L. Davis, K. A. Jørgensen and W.-J. Xiao, Angew. Chem., Int. Ed., 2012, 51, 784 CrossRef CAS PubMed; (b) K. Inamoto, K. Nozawa, M. Yonemoto and Y. Kondo, Chem. Commun., 2011, 47, 11775 RSC; (c) H. Yang, Y. Li, M. Jiang, J. Wang and H. Fu, Chem.–Eur. J., 2011, 17, 5652 CrossRef CAS PubMed; (d) B. Kaboudin, Y. Abedi and T. Yokomatsu, Eur. J. Org. Chem., 2011, 6656 CrossRef CAS; (e) A. D. Chowdhury, S. M. Mobin, S. Mukherjee, S. Bhaduri and G. K. Lahiri, Eur. J. Inorg. Chem., 2011, 3232 CrossRef CAS; (f) J.-M. Xu, X.-Y. Wang, C.-W. Shao, D.-Y. Su, G.-L. Cheng and Y.-F. Hu, Org. Lett., 2010, 12, 1964 CrossRef CAS PubMed.
- H2O2 as oxidant for hydroxylation of arylboronic acids, see: (a) M. Gohain, M. du Plessis, J. H. van Tonder and B. C. B. Bezuidenhoudt, Tetrahedron Lett., 2014, 55, 2082 CrossRef CAS PubMed; (b) L. Wang, D.-Y. Dai, Q. Chen and M.-Y. He, Asian J. Org. Chem., 2013, 2, 1040 CrossRef CAS; (c) N. Mulakayala, Ismail, K. M. Kumar, R. K. Rapolu, B. Kandagatla, P. Rao, S. Oruganti and M. Pal, Tetrahedron Lett., 2012, 53, 6004 CrossRef CAS PubMed; (d) A. Gogoi and U. Bora, Synlett, 2012, 23, 1079–1081 CrossRef CAS PubMed; (e) G. K. S. Prakash, S. Chacko, C. Panja, T. E. Thomas, L. Gurung, G. Rasul, T. Mathew and G. A. Olah, Adv. Synth. Catal., 2009, 351, 1567 CrossRef CAS; (f) J. Simon, S. Salzbrunn, G. K. S. Prakash, N. A. Petasis and G. A. Olah, J. Org. Chem., 2001, 66, 633 CrossRef CAS; (g) H. G. Kuivila and A. G. Armour, J. Am. Chem. Soc., 1957, 79, 5659 CrossRef CAS; (h) M. F. Hawthorne, J. Org. Chem., 1957, 22, 1001 CrossRef CAS; (i) H. G. Kuivila, J. Am. Chem. Soc., 1955, 77, 4014 CrossRef CAS.
- Oxone as oxidant for hydroxylation of arylboronic acids, see: (a) R. E. Maleczka Jr, F. Shi, D. Holmes and M. R. Smith III, J. Am. Chem. Soc., 2003, 125, 7792 CrossRef PubMed; (b) B. R. Travis, B. P. Ciaramitaro and B. Borhan, Eur. J. Org. Chem., 2002, 3429 CrossRef CAS; (c) K. S. Webb and D. Levy, Tetrahedron Lett., 1995, 36, 5117 CrossRef CAS.
- (NH)4S2O8 as oxidant for hydroxylation of arylboronic acids, see: C. A. Contreras-Celedón, L. Chacón-García and N. J. L. Corral, J. Chem., 2014 DOI:10.1155/2014/569572.
- HOF·CH3CN as oxidant for hydroxylation of arylboronic acids, see: J. Gatenyo, I. Vints and S. Rozen, Chem. Commun., 2013, 49, 7379 RSC.
- NaBO3 as oxidant for hydroxylation of arylboronic acids, see: (a) P. Fontani, B. Carboni, M. Vaultier and G. Maas, Synthesis, 1991, 605 CrossRef CAS; (b) E. J. Nanni and R. Ray, J. Am. Chem. Soc., 1980, 102, 7591 CrossRef CAS.
- NaClO2 as oxidant for hydroxylation of arylboronic acids, see: P. Gogoi, P. Bezboruah, J. Gogoi and R. C. Boruah, Eur. J. Org. Chem., 2013, 7291 CrossRef CAS.
- NH2OH as oxidant for hydroxylation of arylboronic acids, see: E. Kianmehr, M. Yahyaee and K. Tabatabai, Tetrahedron Lett., 2007, 48, 2713 CrossRef CAS PubMed.
- TBHP as oxidant for hydroxylation of arylboronic acids, see: S.-M. Guo, L. Lu and H. Cai, Synlett, 2013, 24, 1712 CrossRef CAS PubMed.
- N-oxide as oxidant for hydroxylation of arylboronic acids, see: C. Zhu, R. Wang and J. R. Falck, Org. Lett., 2012, 14, 3494 CrossRef CAS PubMed.
- m-CPBA as oxidant for hydroxylation of arylboronic acids, see: D.-S. Chen and J.-M. Huang, Synlett, 2013, 24, 499–501 CrossRef CAS PubMed.
- (a) W. Q. Wu and H. F. Jiang, Acc. Chem. Res., 2012, 45, 1736 CrossRef CAS PubMed; (b) A. N. Vedernikov, Acc. Chem. Res., 2012, 45, 803 CrossRef CAS PubMed; (c) A. N. Campbell and S. S. Stahl, Acc. Chem. Res., 2012, 45, 851 CrossRef CAS PubMed; (d) Z. Z. Shi, C. Zhang, C. H. Tang and N. Jiao, Chem. Soc. Rev., 2012, 41, 3381 RSC; (e) S. S. Stahl, Science, 2005, 309, 1824 CrossRef CAS PubMed; (f) S. S. Stahl, Angew. Chem., Int. Ed., 2004, 43, 3400 CrossRef CAS PubMed.
- W. Ding, J.-R. Chen, Y.-Q. Zou, S.-W. Duan, L.-Q. Lu and W.-J. Xiao, Org. Chem. Front., 2014, 1, 151 RSC.
- H. Kotoučová, I. Strnadová, M. Kovandová, J. Chudoba, H. Dvořákovác and R. Cibulka, Org. Biomol. Chem., 2014, 12, 2137 Search PubMed.
- (a) Y. Zhong and W. Han, Chem. Commun., 2014, 50, 3874 RSC; (b) Q. Zhou, S. H. Wei and W. Han, J. Org. Chem., 2014, 79, 1454 CrossRef CAS PubMed; (c) L. F. Yao, Q. Zhou, W. Han and S. H. Wei, Eur. J. Org. Chem., 2012, 6856 CrossRef CAS.
- For reviews on PEG, see: (a) E. Colacino, J. Martinez, F. Lamaty, L. S. Patrikeeva, L. L. Khemchyan, V. P. Ananikov and I. P. Beletskaya, Coord. Chem. Rev., 2012, 256, 2893 CrossRef CAS PubMed; (b) J. Chen, S. K. Spear, J. G. Huddleston and R. D. Rogers, Green Chem., 2005, 7, 64 RSC.
- ESI.†.
- An example of photocatalyzed hydroxylation of arylboronic acids, see: S. D. Sawant, A. D. Hudwekar, K. A. Aravinda Kumar, V. Venkateswarlu, P. Pal Singh and R. A. Vishwakarma, Tetrahedron Lett., 2014, 55, 811 CrossRef CAS PubMed.
- A. Berkessel and J. A. Adrio, J. Am. Chem. Soc., 2006, 128, 8421 CrossRef CAS PubMed.
- (a) N. Mulakayala, K. M. Kumar, R. K. Rapolu, B. Kandagatla, P. Rao, S. Oruganti and M. Pal, Tetrahedron Lett., 2012, 53, 6004 CrossRef CAS PubMed; (b) G. W. Kabalka, P. P. Wadgaonkar and T. M. Shoup, Organometallics, 1990, 9, 1316 CrossRef CAS.
- General procedure for aerobic oxidation of aryl/heteroaryl boronic acids to phenols with ambient air: a flask was charged with aryl/heteroaryl boronic acid 1 (0.5 mmol), N2H4·H2O (0.25 mmol, 14.4 μL), Cs2CO3 (1.0 mmol, 329.1 mg), H2O (2.5 mmol, 45.0 μL), and PEG-400 (2.0 g). Then, the flask was stirred at 80 °C in open air for the indicated time. At the end of the reaction, the reaction mixture was acidified with dilute aqueous HCl and extracted with ethyl acetate (3 × 15 mL). The organic phases were combined, and the volatile components were evaporated under reduced pressure. The crude product was purified by column chromatography on silica gel (petroleum ether–ethyl acetate).
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 996231. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra05589f |
| This journal is © The Royal Society of Chemistry 2014 |