
The temperature-dependence of the structure-directing effect of 2-methylpiperazine in the synthesis of open-framework aluminophosphates†
Abstract
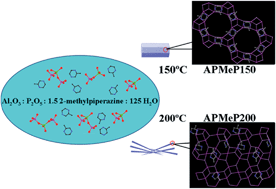
Upon heating the same initial mixture with the composition of Al2O3 : P2O5 : 1.5 2-methylpiperazine : 125H2O at 150 and 200 °C, two layered aluminophosphates were obtained. The crystallization processes of these layered aluminophosphates were investigated using multiple techniques. The change in the pH values of the solutions, the change in the concentrations of Al and P in the liquid phases, and the evolution of the coordination states of Al and P in the solid products were monitored. The “reverse temporal evolution” analyses of each structure showed that the core units or the possible onsets (nuclei) of crystallization of each structure were significantly different from each other. The results showed that the heating temperature altered the structure-directing effect of 2-methylpiperazine and the crystallization process of the initial mixture via changing the structure and chemical properties of the structure-directing agent of 2-methylpiperazine, the equilibrium of the reactions occurring among the source materials, and the assembly of the inorganic oligomers.
 Please wait while we load your content...
Please wait while we load your content...

