
Facile synthesis of ultrasmall TiO2 nanocrystals/porous carbon composites in large quantity and their photocatalytic performance under visible light†
Abstract
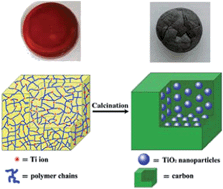
In this paper, we demonstrate a facile and scalable route to the preparation of composites containing ultrasmall TiO2 nanocrystals and porous carbon matrix. In this method, the titanium ions are covalently introduced to polymer chains and transformed into TiO2 nanocrystals directly in solid matrices, which allows the generation of well dispersed TiO2 nanocrystals with small size in the entire carbon matrix. To our knowledge, this is the first time that ultrasmall TiO2 nanocrystals are incorporated into a bulk porous carbon matrix. In comparison with pure TiO2 particles, the composites exhibit significant improvement in photocatalytic degradation of methyl blue under visible light irradiation, which might be attributed to the ultrasmall size of TiO2 nanocrystals as well as the high separation efficiency of photogenerated electrons and holes based on the synergistic effect between TiO2 nanocrystals and carbon matrices. Furthermore, the composites could be easily recycled without obvious activity loss.
 Please wait while we load your content...
Please wait while we load your content...

