
Design, synthesis and biological evaluation of 4′-demethyl-4-deoxypodophyllotoxin derivatives as novel tubulin and histone deacetylase dual inhibitors†
Xuan Zhang‡
a,
Jie Zhang‡b,
Mingbo Suc,
Yubo Zhouc,
Yi Chen*b,
Jia Li*c and
Wei Lu*a
aInstitute of Drug Discovery and Development, Shanghai Engineering Research Center of Molecular Therapeutics and New Drug Development, East China Normal University, 3663 North Zhongshan Road, Shanghai 200062, PR China. E-mail: wlu@chem.ecnu.edu.cn; Fax: +86(21)62602475
bState Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, PR. China. E-mail: ychen@jding.dhs.org
cNational Center for Drug Screening, State Key Laboratory of Drug Research, Shanghai Institute of Materia Medica, Chinese Academy of Sciences, Shanghai 201203, PR China. E-mail: jli@simm.ac.cn
First published on 22nd August 2014
Abstract
A new class of 4′-demethyl-4-deoxypodophyllotoxin derivatives has been designed and synthesized as tubulin–HDAC dual inhibitors. Biological evaluation of these hybrids included the inhibitory activity of HDAC, in vitro cell cycle analysis in HCT-116 cells as well as cytotoxicity against two cancer cell lines (A549 and HCT116). The distance and angle between the HDAC capping group and the zinc binding group were systematically varied. Compounds 14a and 14c showed most potent dual inhibitory activity and powerful antiproliferative activity on HCT116 and A549 cell lines.
Introduction
According to the World Cancer Report 2014, there were 14 million new cancer cases in 2012 globally and the number is expected to continuously increase within the next two decades.1 The development of cancer pharmacotherapy has a history of more than 70 years. Traditional anti-cancer drugs including cytotoxic agents and endocrine medications have been widely used in a variety of cancer chemotherapy treatments. But the usage of these drugs is always hindered by severe toxicity and other undesirable side effects. The development of targeted molecularly anti-cancer drugs has made significant achievement over the past decade. However, only part of the patients show positive response. In addition, the acquired drug resistance always limits the use of these agents. Diseases with linear pathways might be well treated with single target agents. Cancer is a disease with complex signaling networks. For this reason, it is difficult to treat cancer by using classical targeted drugs alone.2Anti-tumor drug combination therapy can block several key signaling pathways and create synergistic antitumor effects.3–11 This kind of therapeutic regimens can improve therapeutic efficacy and simultaneously reduce drug toxicity. The major defects of combination therapy are the complicated pharmacokinetics and the adverse effects associated with drug interactions.
Recently, the design of multi-target drugs has become a new strategy to overcome these limitations.12–14 These agents can create synergistic anti-cancer effects and exhibit simpler pharmacokinetics. Histone deacetylase (HDAC) involved multi-target drug design is one of the hotspots in this area. HDAC plays an important role in the regulation of gene expression. HDAC inhibitors can cause growth arrest, differentiation, and apoptosis in cancer cells.15–18 Up to date, two HDAC inhibitors, vorinostat (SAHA) and romidepsin have been approved by FDA for the treatment of cutaneous T-cell lymphoma (CTCL).19–21 In general, a HDAC inhibitor consists of a capping group, a zinc-binding group (ZBG) and an appropriate linker (Fig. 1). The simple SAR together with the effectiveness in oncotherapy have attracted many oncologists into the exploration of HDAC-involved multi-target agents.22–28
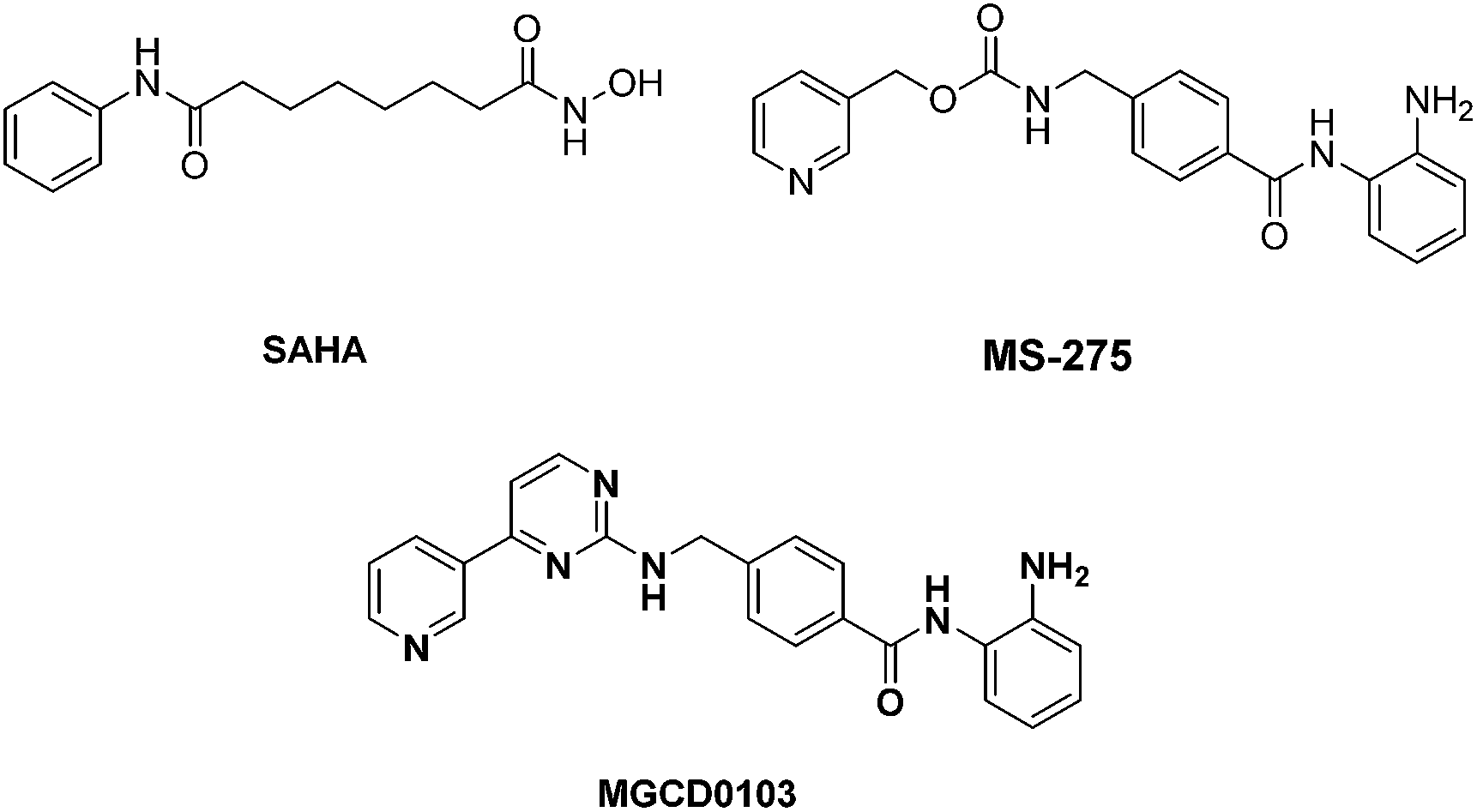 | ||
| Fig. 1 Representative structures of HDAC inhibitors. | ||
HDAC inhibitors can have a synergistic antitumor effect when combined with tubulin inhibitors.29–31 Previously, we disclosed a new class of podophyllotoxin (PPT) derivatives as topoisomerase II (Topo II)–HDAC dual inhibitors.32 Among this series of hybrids, compound 1 displayed the best anti-HDAC activity and was 10- to 20 fold more potent than SAHA, which suggested that the PPT moiety might be a suitable capping group for HDAC inhibitors.
The structure of PPT was first elucidated in 1932 and hundreds of derivatives have been reported. Three semi-synthetic analogues, etoposide (VP-16), teniposide and etopophos have been approved to use in chemotherapy (Fig. 3).33,34 Interestingly, these semi-synthetic derivatives exert their antiproliferative activity through the inhibition of Topo II, which is different from the parent compound PPT. 4-Deoxypodophyllotoxin (DPT) is a PPT analogue, which can inhibit tubulin polymerization and cause cell cycle arrest at G2/M followed by apoptosis.35–37 The structural modifications of DPT are focused on the 4′-position,38–43 the representative compound 4′-demethyl-4-deoxypodophyllotoxin (DDPT) has a comparable antitumor activity with DPT.
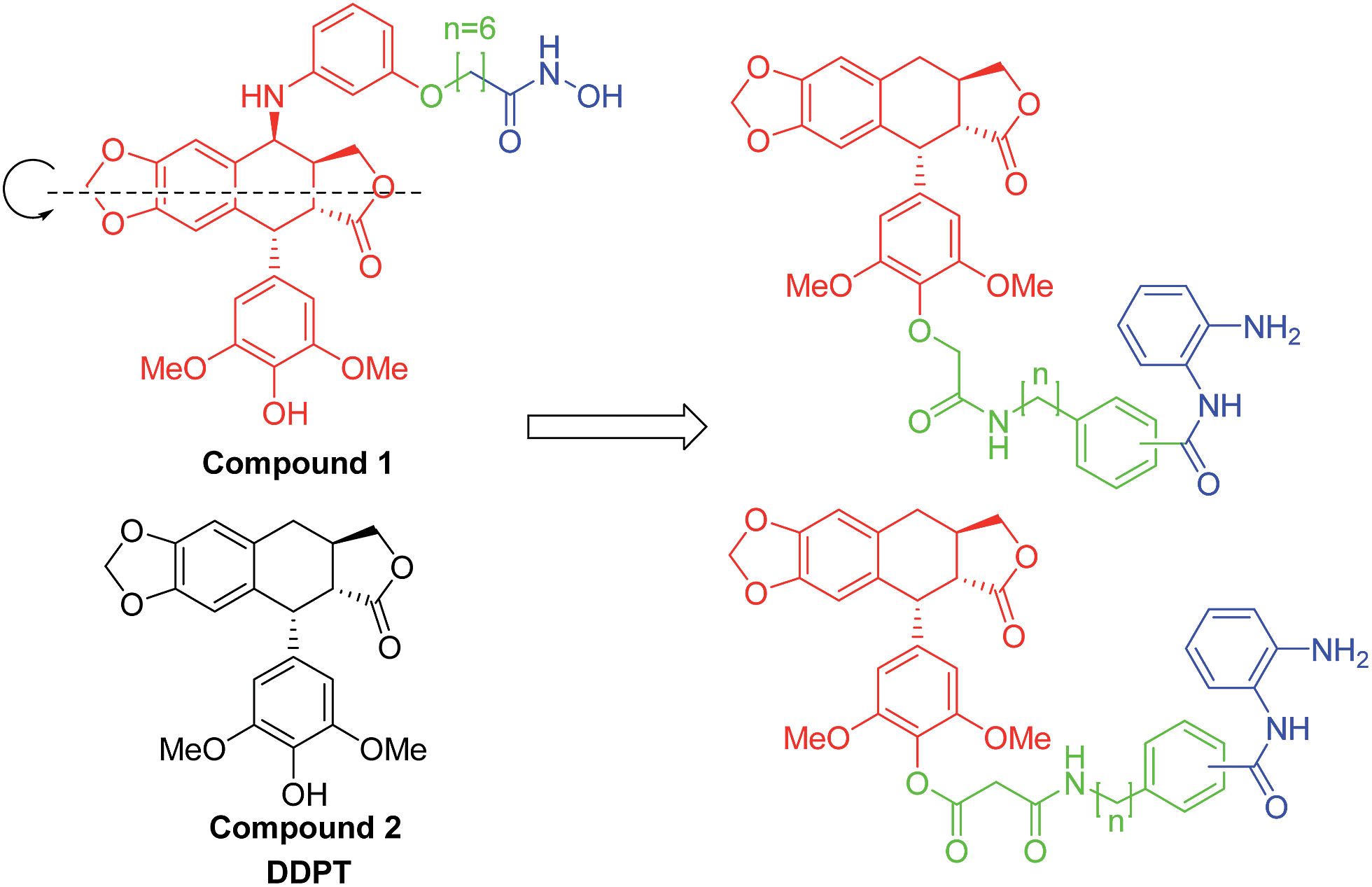 | ||
| Fig. 2 Design of tubulin–HDAC dual inhibitors. | ||
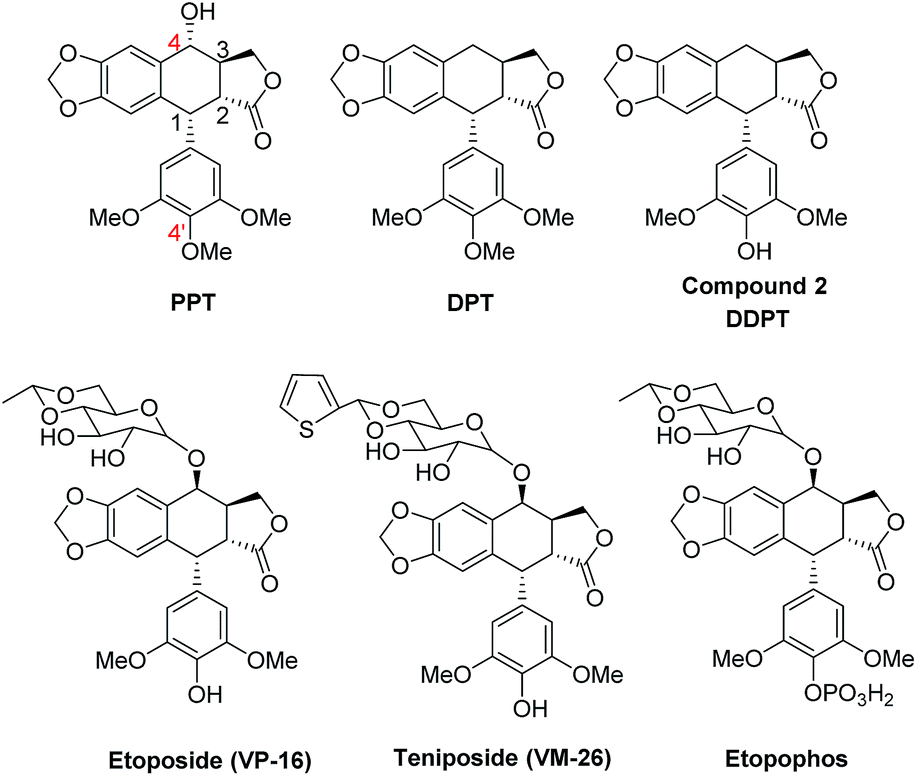 | ||
| Fig. 3 Representative structures of PPT analogues. | ||
As shown in Fig. 2, compound 2 can be regarded as an axial symmetry structure. Therefore, we envisioned that the transplant of HDAC pharmacophore from the 4-position to the 4′-position can achieve a new series of tubulin–HDAC dual inhibitors. Aromatic capping group connection, linker length and ZBG connection were systematically varied to regulate the distance and angle between the cap and ZBG. Biological evaluation of these hybrids includes their anti-HDAC activities, in vitro cell cycle analysis in HCT-116 cells and their antiproliferative activities in two cancer cell lines.
Results and discussion
Chemistry
The general route used for the synthesis of the ether series is depicted in Scheme 1. According to the reported literature, DDPT can be easily prepared from PPT in three steps.43 Treatment of DDPT with tert-butyl 2-bromoacetate followed by removal of the protecting group gave the key intermediate 3 of ether series. Ester series intermediate 4 was prepared through treatment of DDPT with 2,2-dimethyl-1,3-dioxane-4,6-dione.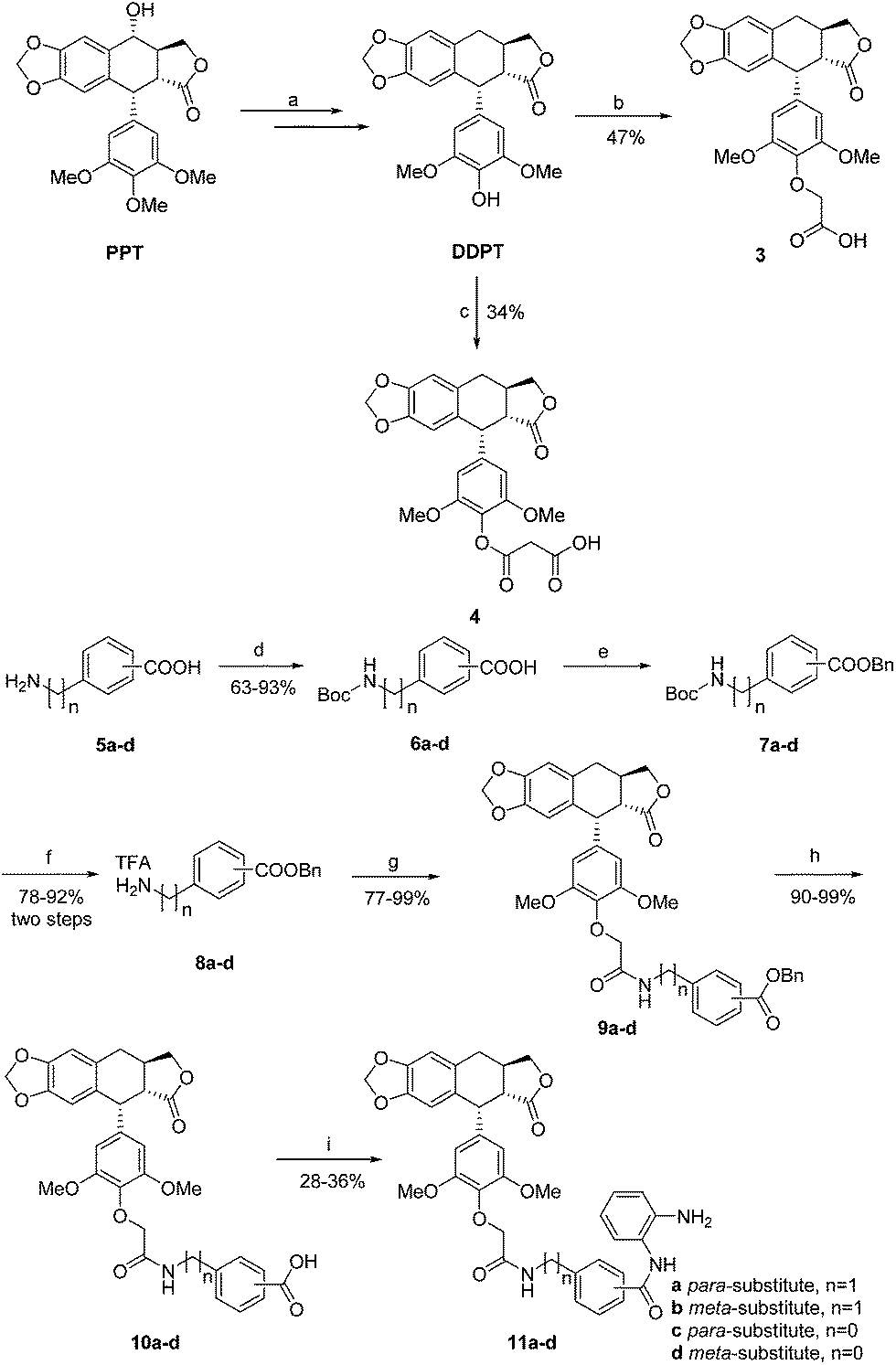 | ||
| Scheme 1 Synthesis of ether series hybrids. Reagents and conditions: (a) TMSI, DCM; BaCO3, acetone, H2O; Pd/C, H2, AcOH, 95 °C; (b) tert-butyl 2-bromoacetate, K2CO3, KI; CF3COOH, DCM; (c) 2,2-dimethyl-1,3-dioxane-4,6-dione, toluene, reflux; (d) Boc2O, NaHCO3, dioxane, H2O; (e) BnBr, Cs2CO3, DMF; (f) TFA, DCM; (g) compound 3, HATU, DIPEA, DCM; (h) Pd(OH)2, H2, MeOH, ethyl acetate; (i) HATU, DIPEA, DCM. | ||
On the other hand, starting from amino acids 5a–d, amines 8a–d can be synthesized smoothly in three steps. Subsequent amidation of acid 3 with the corresponding amine afforded compounds 9a–d. Hydrogenation of these compounds using Pd(OH)2 under hydrogen atmosphere led to benzoic acids 10a–d. Finally, the ether series hybrids 11a–d were obtained after amidation.
Similarly, as shown in Scheme 2, preparation of ester series hybrids 14a–d was started from the amidation of compound 4 and amines 8a–d. Subsequently, esters 12a–d were treated with Pd(OH)2 under hydrogen atmosphere to afford acids 13a–d. Amidation of these acids and 1,2-benzenediamine gave designed hybrids 14a–d.
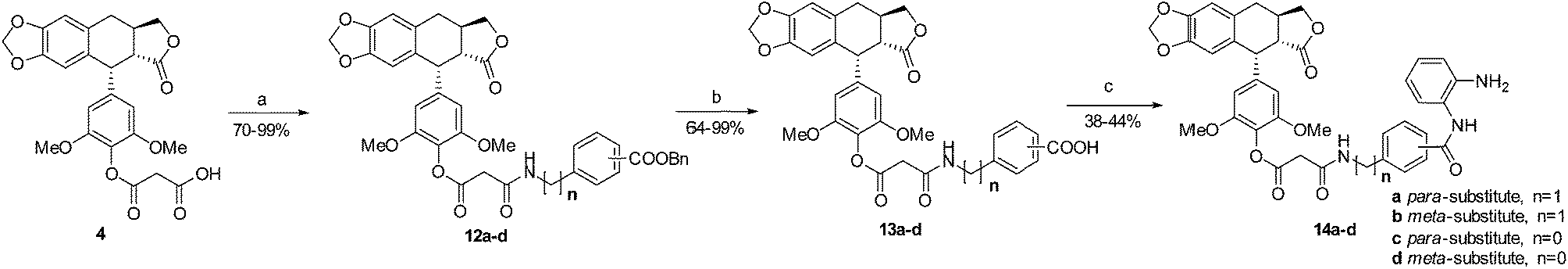 | ||
| Scheme 2 Synthesis of ester series hybrids. Reagents and conditions: (a) HATU, DIPEA, DCM; (b) Pd(OH)2, H2, MeOH, ethyl acetate; (c) HATU, DIPEA, DCM. | ||
Biological assays
| Compound | IC50 [μM] ± SD | ||
|---|---|---|---|
| HDAC-1 | HDAC-2 | HDAC-3 | |
| a Each value was reproduced in three experiments.b IC50 > 20 μg mL−1. | |||
| MGCD0103 | 0.95 ± 0.16 | 0.28 ± 0.02 | 1.67 ± 0.01 |
| Etoposide | —b | — | — |
| DDPT | — | — | — |
| 11a | 15.10 ± 6.17 | 1.69 ± 0.11 | 6.87 ± 2.12 |
| 11b | — | 35.43 ± 5.46 | — |
| 11c | — | 7.51 ± 0.32 | — |
| 11d | — | — | — |
| 14a | 11.09 ± 0.90 | 1.73 ± 0.21 | 11.00 ± 0.80 |
| 14b | 40.71 ± 6.49 | 18.58 ± 4.20 | 30.56 ± 11.99 |
| 14c | 10.96 ± 3.45 | 0.75 ± 0.05 | 5.30 ± 1.15 |
| 14d | — | 13.45 ± 1.71 | 44.11 ± 3.69 |
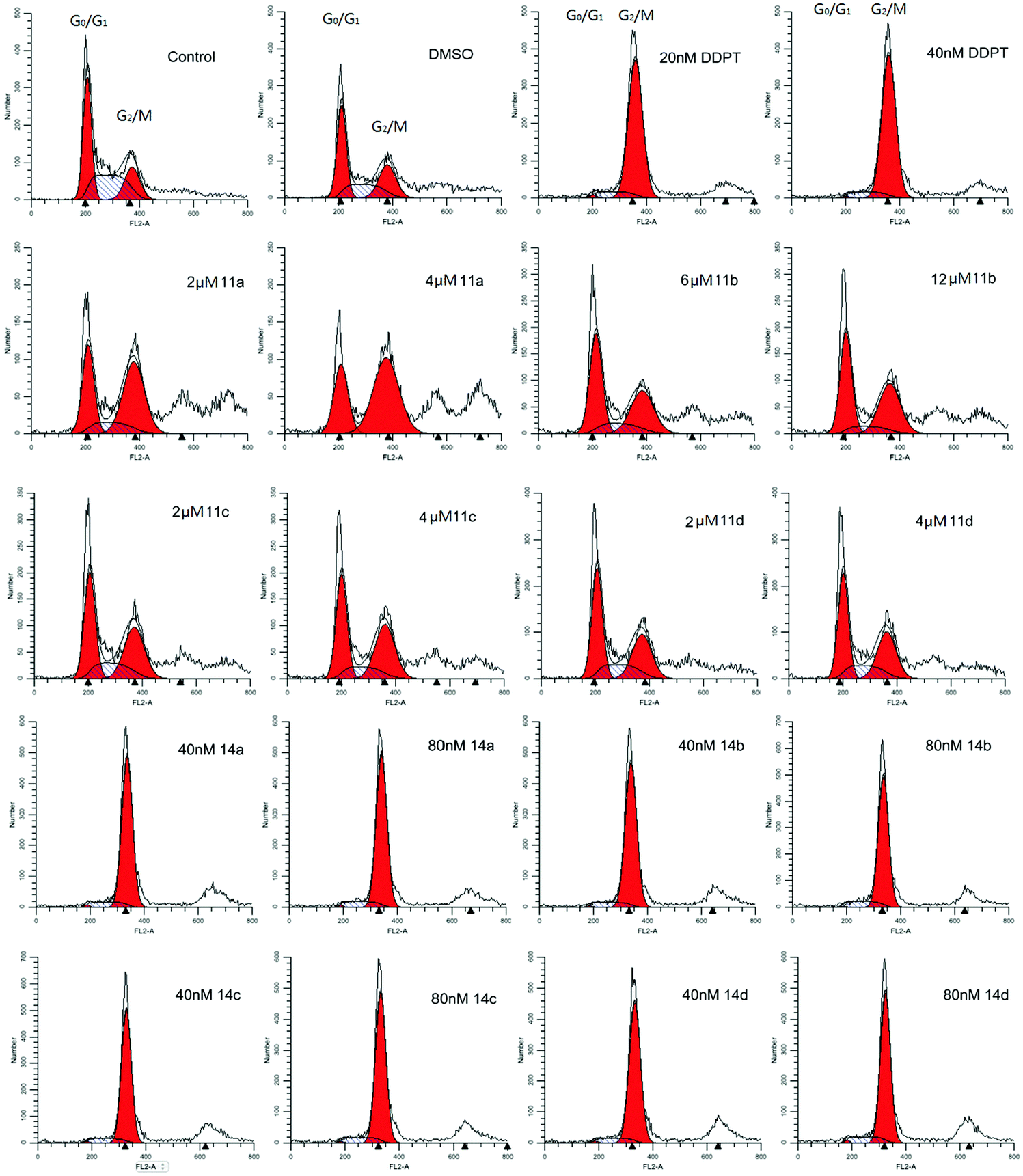 | ||
| Fig. 4 In vitro cell cycle analysis of 11a–d and 14a–d in HCT-116 cells. | ||
| Compound | HCT116 IC50 [μM] ± SD | A549 IC50 [μM] ± SD |
|---|---|---|
| a Each value was reproduced in three experiments. | ||
| MGCD0103 | 1.572 ± 0.349 | 1.645 ± 0.235 |
| Etoposide | 0.873 ± 0.171 | 1.714 ± 0.165 |
| DDPT | 0.016 ± 0.000 | 0.011 ± 0.003 |
| 11a | 1.583 ± 0.414 | 3.789 ± 0.409 |
| 11b | 5.889 ± 0.254 | 2.692 ± 0.315 |
| 11c | 1.600 ± 0.560 | 1.229 ± 0.030 |
| 11d | 1.300 ± 0.770 | 1.033 ± 0.146 |
| 14a | 0.037 ± 0.001 | 0.036 ± 0.001 |
| 14b | 0.036 ± 0.001 | 0.019 ± 0.003 |
| 14c | 0.040 ± 0.001 | 0.037 ± 0.001 |
| 14d | 0.036 ± 0.002 | 0.040 ± 0.002 |
Conclusions
A new class of DDPT derivatives was designed and synthesized as tubulin–HDAC dual inhibitors. Overall, the ester series hybrids showed better HDAC inhibitory activity and tubulin activity than the ether series. ZBG connection had an influence on HDAC inhibitory activity, the para-substituted molecules exhibited better anti-HDAC activity than the meta-substituted one. On the other hand, hybrids connected with ester bond displayed better anti-tubulin activity than the than the ether series hybrids. Compounds 14a and 14c acted as potent tubulin–HDAC dual inhibitors and showed good antiproliferative activity against A549 and HCT116 cell lines. The present encouraging results give a promising future optimization of the series.Acknowledgements
This work was supported by the Youth Scientific Innovation Foundation of East China Normal University (no. 78210157 and no. 78210198), the State Key Laboratory of Drug Research (no. SIMM1203KF-10), the grants of The National Natural Science Foundation of China (no. 81172936, no. 81173033 and no. 21102046), and grants of The Fundamental Research Funds for the Central Universities.Notes and references
- International Agency for Research on Cancer, World Health Organization: Geneva, 2014.
- L. Costantino and D. Barlocco, Future Med. Chem., 2013, 5, 5–7 CrossRef CAS PubMed.
- M. S. Kim, M. Blake, J. H. Baek, G. Kohlhagen, Y. Pommier and F. Carrier, Cancer Res., 2003, 63, 7291–7300 CAS.
- F. Bruzzese, M. Rocco, S. Castelli, E. Di Gennaro, A. Desideri and A. Budillon, Mol. Cancer Ther., 2009, 8, 3075–3087 CrossRef CAS PubMed.
- R. L. Bevins and S. G. Zimmer, Cancer Res., 2005, 65, 6957–6966 CrossRef CAS PubMed.
- M. Ocker, A. Alajati, M. Ganslmayer, S. Zopf, M. Luders, D. Neureiter, E. G. Hahn, D. Schuppan and C. Herold, J. Cancer Res. Clin. Oncol., 2005, 131, 385–394 CrossRef CAS PubMed.
- B. Sarcar, S. Kahali and P. Chinnaiyan, J. Neuro-Oncol., 2010, 99, 201–207 CrossRef CAS PubMed.
- M. Dokmanovic, C. Clarke and P. A. Marks, Mol. Cancer Res., 2007, 5, 981–989 CrossRef CAS PubMed.
- H. Y. Lin, C. S. Chen, S. P. Lin, J. R. Weng and C. S. Chen, Med. Res. Rev., 2006, 26, 397–413 CrossRef CAS PubMed.
- M. J. Lee, Y. S. Kim, S. Kummar, G. Giaccone and J. B. Trepel, Curr. Opin. Oncol., 2008, 20, 639–649 CrossRef CAS PubMed.
- C. S. Chen, S. C. Weng, P. H. Tseng, H. P. Lin and C. S. Chen, J. Biol. Chem., 2005, 280, 38879–38887 CrossRef CAS PubMed.
- K. Nepali, S. Sharma, M. Sharma, P. Bedi and K. Dhar, Eur. J. Med. Chem., 2014, 77, 422–487 CrossRef CAS PubMed.
- K. A. Papavassiliou and A. G. Papavassiliou, Expert Opin. Invest. Drugs, 2013, 1–4 Search PubMed.
- H. Wang and B. W. Dymock, Expert Opin. Ther. Pat., 2009, 19, 1727–1757 CrossRef CAS PubMed.
- A. Saito, T. Yamashita, Y. Mariko, Y. Nosaka, K. Tsuchiya, T. Ando, T. Suzuki, T. Tsuruo and O. Nakanishi, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 4592–4597 CrossRef CAS.
- R. D. Glick, S. L. Swendeman, D. C. Coffey, R. A. Rifkind, P. A. Marks, V. M. Richon and M. P. La Quaglia, Cancer Res., 1999, 59, 4392–4399 CAS.
- L. M. Butler, D. B. Agus, H. I. Scher, B. Higgins, A. Rose, C. Cordon-Cardo, H. T. Thaler, R. A. Rifkind, P. A. Marks and V. M. Richon, Cancer Res., 2000, 60, 5165–5170 CAS.
- A. K. Oyelere, P. C. Chen, W. Guerrant, S. C. Mwakwari, R. Hood, Y. Zhang and Y. Fan, J. Med. Chem., 2009, 52, 456–468 CrossRef CAS PubMed.
- B. S. Mann, J. R. Johnson, M. H. Cohen, R. Justice and R. Pazdur, Oncologist, 2007, 12, 1247–1252 CrossRef CAS PubMed.
- C. Grant, F. Rahman, R. Piekarz, C. Peer, R. Frye, R. W. Robey, E. R. Gardner, W. D. Figg and S. E. Bates, Expert Rev. Anticancer Ther., 2010, 10, 997–1008 CrossRef CAS PubMed.
- S. C. Mwakwari, V. Patil, W. Guerrant and A. K. Oyelere, Curr. Top. Med. Chem., 2010, 10, 1423–1440 CrossRef CAS.
- X. Cai, H. X. Zhai, J. Wang, J. Forrester, H. Qu, L. Yin, C. J. Lai, R. Bao and C. Qian, J. Med. Chem., 2010, 53, 2000–2009 CrossRef CAS PubMed.
- C. J. Lai, R. Bao, X. Tao, J. Wang, R. Atoyan, H. Qu, D. G. Wang, L. Yin, M. Samson, J. Forrester, B. Zifcak, G. X. Xu, S. DellaRocca, H. X. Zhai, X. Cai, W. E. Munger, M. Keegan, C. V. Pepicelli and C. Qian, Cancer Res., 2010, 70, 3647–3656 CrossRef CAS PubMed.
- S. Mahboobi, S. Dove, A. Sellmer, M. Winkler, E. Eichhorn, H. Pongratz, T. Ciossek, T. Baer, T. Maier and T. Beckers, J. Med. Chem., 2009, 52, 2265–2279 CrossRef CAS PubMed.
- S. Mahboobi, A. Sellmer, M. Winkler, E. Eichhorn, H. Pongratz, T. Ciossek, T. Baer, T. Maier and T. Beckers, J. Med. Chem., 2010, 53, 8546–8555 CrossRef CAS PubMed.
- F. Thaler, Pharm. Pat. Anal., 2012, 1, 75–90 CrossRef CAS PubMed.
- X. Zhang, M. Su, Y. Chen, J. Li and W. Lu, Molecules, 2013, 18, 6491–6503 CrossRef CAS PubMed.
- X. Zhang, J. Zhang, L. J. Tong, Y. Luo, M. B. Su, Y. Zang, J. Li, W. Lu and Y. Chen, Bioorg. Med. Chem., 2013, 21, 3240–3244 CrossRef CAS PubMed.
- J. H. Kim, E. K. Yoon, H. J. Chung, S. Y. Park, K. M. Hong, C. H. Lee, Y. S. Lee, K. Choi, Y. Yang, K. Kim and I. H. Kim, Apoptosis, 2013, 18, 110–120 CrossRef CAS PubMed.
- N. H. Chobanian, V. L. Greenberg, J. M. Gass, C. P. Desimone, J. R. Van Nagell and S. G. Zimmer, Anticancer Res., 2004, 24, 539–545 CAS.
- V. Zuco, M. De Cesare, R. Cincinelli, R. Nannei, C. Pisano, N. Zaffaroni and F. Zunino, PLoS One, 2011, 6, e29085 CAS.
- X. Zhang, B. Bao, X. Yu, L. Tong, Y. Luo, Q. Huang, M. Su, L. Sheng, J. Li, H. Zhu, B. Yang, X. Zhang, Y. Chen and W. Lu, Bioorg. Med. Chem., 2013, 21, 6981–6995 CrossRef CAS PubMed.
- K. R. Hande, Eur. J. Cancer, 1998, 34, 1514–1521 CrossRef CAS.
- K. R. Hande, Biochim. Biophys. Acta, 1998, 1400, 173–184 CrossRef CAS.
- S.-J. Suh, J.-R. Kim, U.-H. Jin, H.-S. Choi, Y.-C. Chang, Y.-C. Lee, S.-H. Kim, I.-S. Lee, T. C. Moon and H.-W. Chang, Vasc. Pharmacol., 2009, 51, 13–20 CrossRef CAS PubMed.
- Y. Yong, S. Y. Shin, Y. H. Lee and Y. Lim, Bioorg. Med. Chem. Lett., 2009, 19, 4367–4371 CrossRef CAS PubMed.
- S. Y. Shin, Y. Yong, C. G. Kim, Y. H. Lee and Y. Lim, Cancer Lett., 2010, 287, 231–239 CrossRef CAS PubMed.
- Z. W. Zhang, J. Q. Zhang, L. Hui, S. W. Chen and X. Tian, Eur. J. Med. Chem., 2010, 45, 1673–1677 CrossRef CAS PubMed.
- Y. Kim, Y.-J. You, N.-H. Nam and B.-Z. Ahn, Bioorg. Med. Chem. Lett., 2002, 12, 3435–3438 CrossRef CAS.
- Y.-J. You, Y. Kim, N.-H. Nam, S.-C. Bang and B.-Z. Ahn, Eur. J. Med. Chem., 2004, 39, 189–193 CrossRef CAS PubMed.
- Y. Jin, J. Liu, W. T. Huang, S. W. Chen and L. Hui, Eur. J. Med. Chem., 2011, 46, 4056–4061 CrossRef CAS PubMed.
- H. Xu and J.-J. Wang, Bioorg. Med. Chem. Lett., 2010, 20, 2500–2502 CrossRef CAS PubMed.
- S.-W. Chen, Y.-Y. Gao, N.-N. Zhou, J. Liu, W.-T. Huang, L. Hui, Y. Jin and Y.-X. Jin, Bioorg. Med. Chem. Lett., 2011, 21, 7355–7358 CrossRef CAS PubMed.
- N. Zhou, O. Moradei, S. Raeppel, S. Leit, S. Frechette, F. Gaudette, I. Paquin, N. Bernstein, G. Bouchain and A. Vaisburg, J. Med. Chem., 2008, 51, 4072–4075 CrossRef CAS PubMed.
- M. Jordan, Curr. Med. Chem.: Anti-Cancer Agents, 2002, 2, 1–17 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of experimental procedure, spectral data of all novel compounds. See DOI: 10.1039/c4ra05508j |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2014 |