
High electrocatalytic performance of platinum and manganese dioxide nanoparticle decorated reduced graphene oxide sheets for methanol electro-oxidation†
Abstract
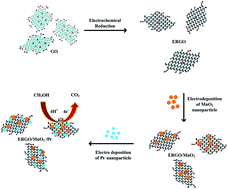
In this study we report the synthesis of a novel Pt–MnO2–ERGO electrocatalyst by the deposition of MnO2 and Pt nanoparticles decorated on reduced graphene oxide sheets using a simple electrochemical method. The as prepared MnO2 and Pt nanoparticles decorated on the reduced graphene oxide sheets (Pt–MnO2–ERGO electrocatalysts) were characterized by scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), energy-dispersive X-ray spectroscopy (EDX) and X-ray photoelectron spectroscopy (XPS). The cyclic voltammetric (CV), chronoamperometric and electrochemical impedance spectroscopic (EIS) measurements show high electrocatalytic activity and stability of the electrodes towards the methanol oxidation reaction in nitrogen saturated sulfuric acid aqueous solutions and in mixed sulfuric acid and methanol aqueous solutions. The voltammetric results show the electrocatalytic characteristics of the Pt–MnO2–ERGO electrocatalysts, which exhibit superior electrocatalytic activity (including good poison tolerance, and low onset potential) and stability toward electro-oxidation of methanol in a model reaction. The electrochemical impedance spectroscopic result shows good electrocatalytic activity in relation to methanol oxidation and improved tolerance of CO. In addition, the as designed Pt–MnO2–ERGO nanocomposite modified electrode with a novel structure can be directly employed for fuel cells.
 Please wait while we load your content...
Please wait while we load your content...

