
Neutral Aun (n = 3–10) clusters catalyze acetylene hydrochlorination: a density functional theory study†
Yang Wang,
Mingyuan Zhu,
Lihua Kang* and
Bin Dai*
College of Chemistry and Chemical Engineering/Key Laboratory for Green Processing of Chemical Engineering of Xinjiang Bingtuan. Shihezi University, Shihezi, Xinjiang 832000, PR China. E-mail: kanglihua@shzu.edu.cn; Fax: +86-0993-2057270; Tel: +86-0993-2057213
First published on 18th August 2014
Abstract
The mechanisms of acetylene hydrochlorination to vinyl chloride catalyzed by neutral Au3–10 clusters were systematically investigated using density functional theory with the B3LYP/LANL2DZ function. In this reaction, the gold cluster functions as a bridge of electron transfer: the electrons transfer from C2H2 to the gold cluster then from the gold cluster to HCl. HCl and C2H2 are simultaneously activated by the gold cluster, which presents a synergistic effect in co-adsorption. In the size range of Au3 to Au10, all gold clusters undergo the same catalytic cycle. The whole process of the acetylene hydrochlorination on the gold cluster consists of two transition states and one intermediate, and the dissociation of hydrogen chloride is the rate-controlling step. Overall, small-sized gold clusters perform better than large-sized clusters, and the odd-number atom clusters are better than even-number atom clusters.
1. Introduction
Interest in gold catalysis has been increasing, and the subject is now well-established in both heterogeneously and homogeneously catalyzed reactions.1–6 Gold, in its bulk form, is known to possess little or no catalytic activity. However, small gold clusters exhibit remarkably different fundamental properties. An increasing number of studies have shown that gold clusters with appropriate sizes show considerably good activity for various catalytic reactions.7–30 For example, nanosized gold clusters exhibit a high catalytic activity toward carbon monoxide oxidation,8,17–19 selective oxidation of olefin and alcohol,10,11 water–gas shift reaction,12,14 synthesis of hydrogen peroxide,13 S–H bond rupture of thiophenol and mercaptan,20,21 acetylene hydrogenation,16 and acetylene hydrochlorination.7,9,15,26–30 Among various reactions catalyzed by gold clusters, acetylene hydrochlorination has received the most attention because of its considerable economic benefits, wide range of industrial applications, and huge potential for improvement.In 1985, Hutchings et al. predicted that supported gold catalyst may be a substitute for mercuric chloride as the more “green material” for the hydrochlorination of acetylene.7 Since then, AuCl3 was found to be the best catalyst for acetylene hydrochlorination from among 30 carbon-supported metal chlorides.23 In 2013, L. Kang et al. studied the reaction mechanism of C2H3Cl over an MClx (M = Hg, Au, Ru; x = 2, 3) catalyst, and they confirmed the conclusion theoretically.24 However, AuCl3 catalyst was unstable due to deactivation, and J. Zhang et al. have studied the deactivation mechanism over AuCl3 dimer model catalyst using DFT.25 In 2014, M. Zhu and his coworkers developed different methods to inhibit the deactivation of Au3+ like adding 1, 10-phenanthroline or polypyrrole (PPy) into AuCl3 catalyst.26–29 Given the improvement in catalyst preparation, A. Corma et al. reported a method to prepare isolated gold atoms supported on functionalized carbon nanotubes,21 which shows that the ultra-fine nanoparticle is not only limited in theoretical research. A. Corma et al. found that single gold atoms are not active, but they aggregate under reaction conditions into gold clusters of low atomicity that exhibit a catalytic activity comparable to that of sulfhydryl oxidase enzymes. When clusters grow into larger nanoparticles, catalyst activity drops to zero. To improve the utilization efficiency of Au catalyst for the reduction of 4-nitrophenol, X. Jia et al. prepared triangular Au nanoplates on functional reduced graphene oxide by a facile method. The products with ultra-low trace amounts of Au afforded high catalytic efficiency.22 Even though numerous studies have shown that properly sized nano-gold clusters exhibit high catalytic activities for various reactions,7–22 no investigations have been conducted thus far on the acetylene hydrochlorination reaction by using neutral gold catalysts. We believe that theoretical calculations should precede experimental investigations and provide direction for experimental studies. The precise control of the crystal plane and cluster size of gold nanoparticles in experiments is difficult, but controlling these parameters is easily achievable in theoretical calculations.
In this paper, we present a DFT study on the acetylene hydrochlorination of a neutral Au cluster in an attempt to answer the following questions: (1) where and how are HCl and C2H2 activated on an Au cluster? (2) Where and how does acetylene hydrochlorination occur? (3) What is the catalytic function of the Au cluster? Several possible reaction pathways are explored with the aim of unraveling the details of acetylene hydrochlorination on an Aun cluster. We predict that a deep understanding of these details will be important in this field of study.
2. Computational methods
All density functional calculations were performed using the Guassian09 program package.31 No geometric constraints were assumed in geometry optimization. All structures containing Au were optimized and characterized at B3LYP/LANL2DZ. The nonlocal correlation functional of Lee, Yang, and Parr32 (B3LYP) with the 6-31++G** basis set was used for H, C, and Cl atoms,33 and the Los Alamos effective core pseudo-potentials (ECP) basis set LANL2DZ was used for the Au atoms. All reported charges are Mulliken local charges (Au). The relative energies of the reactants, products, intermediates, and transition states presented in this study were zero-point-energy (ZPE) obtained from frequency calculations at the same level of optimization. All stationary points were characterized as the minima (no imaginary frequency) or transition state (one imaginary frequency) via Hessian calculation. Intrinsic reaction coordinate (IRC) calculations34,35 were performed to determine if each transition state links the correct product with the reactant. Basis set superposition error (BSSE) corrections evaluated by the counterpoise method36 were taken into account.Through the frontier molecular orbital (FMO) and charge distribution analysis, we can determine the approximate distribution of the active sites of the Aun cluster, and the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energies of the neutral Aun clusters, C2H2, and HCl were theoretically calculated at the B3LYP/LANL2DZ. We then calculated the HOMO–LUMO energy gaps as two groups, namely, between HOMO (gold clusters) and LUMO (C2H2/HCl), and between LUMO (gold clusters) and HOMO (C2H2/HCl). We obtained the lowest HOMO–LUMO energy gaps and confirmed the electron donor and acceptor. Thus, we can know which FMO factors prominently in the adsorption processes. Besides, through the charge distribution analysis of isolated cluster and molecules in the adsorbed complexes, that can further verify the predicted result of FMO analysis, which makes our results be more persuasive and reliable.
An important reference point for this calculation is the adsorption energy for HCl and C2H2 adsorbed on isolated Aun clusters. In this paper, we used the following definitions for adsorption energy.
When HCl or C2H2 is adsorbed on isolated Au, the adsorption energy is calculated as:
| E = E (system) −E (Au) −E (HCl–C2H2) |
When HCl is adsorbed on Au–C2H2, the adsorption energy is calculated as:
| E = E (system) −E (Au–C2H2) −E (HCl) |
When C2H2 is adsorbed on Au–HCl, the adsorption energy is calculated as:
| E = E (system) −E (Au–HCl) −E (C2H2) |
E (system) is the total energy of adsorption system; E (Au–C2H2) and E (Au–HCl) denote the energy of the Au complexes that absorbs C2H2 and HCl, respectively.
3. Results and discussion
3.1 Co-adsorption of C2H2 and HCl on Aun (n = 3–10) clusters
In Fig. S3,† we compared the energy gaps of HOMO–LUMO (HCl → Aun) and HOMO–LUMO (C2H2 → Aun) and yielded the following two main results: (1) for both HCl and C2H2, the energy gaps of even-numbered Aun clusters are larger than those on adjacent odd-numbered ones, and (2) overall, the energy gaps of HOMO–LUMO (HCl → Aun) are larger than those of HOMO–LUMO (C2H2 → Aun) for even-numbered ones. The energy gaps denote the ability of the electrons to transfer between HCl/C2H2 and Aun clusters. To explore the electronic properties and the interactions between Aun–C2H2 complexes and HCl, we calculated the FMO and corresponding orbital energies of the Aun–C2H2 complexes, as shown in Fig. 1. In Table 1, we listed the orbital energies of the LUMO and HOMO of HCl and Aun–C2H2 and their different energy gaps. The energy gaps of HOMO–LUMO (Aun–C2H2 → HCl) are smaller than those of HOMO–LUMO (HCl → Aun–C2H2). This result indicates that Aun–C2H2 complexes are electron donors and HCl is an electron acceptor. The electrons transfer from the HOMO of Aun–C2H2 complexes (almost from the Aun clusters) to the LUMO of HCl. Therefore, we can preliminarily predict the distribution of reaction sites from the HOMO of Aun–C2H2 complexes in Fig. 1. The following points are noted: Au3–5–C2H2 complexes only have one adsorption site for HCl; Au7–C2H2 complex has two adsorption sites, namely, Au (1) and Au (4) atoms (the code numbers of Au clusters are shown in Fig. S1†); in Au6,8,9–C2H2 complexes, the three co-adsorption configurations are different from the others because C2H2 was adsorbed vertically on Au6,8,9. Their adsorption sites are a block of void that comprised three Au atoms. For example, the adsorption void of the Au6,8–C2H2 complex is composed of Au (1), Au (2), and Au (4), whereas that of the Au9–C2H2 complex consists of Au (3), Au (2), and Au (5). | ||
| Fig. 1 Frontier molecular orbitals and corresponding orbital energies of the Aun–C2H2 (n = 3–10) calculated at the B3LYP/LANL2DZ level of theory (energies in eV, isovalue = 0.02). | ||
| LUMO | HOMO | HOMO–LUMO (HCl → Aun–C2H2) | HOMO–LUMO (Aun–C2H2 → HCl) | |
|---|---|---|---|---|
| HCl | −0.81 | −9.19 | ||
| Au3–C2H2 | −3.57 | −5.00 | 5.62 | 4.19 |
| Au4–C2H2 | −2.98 | −5.96 | 6.21 | 5.14 |
| Au5–C2H2 | −3.95 | −5.24 | 5.24 | 4.42 |
| Au6–C2H2 | −2.94 | −6.29 | 6.25 | 5.48 |
| Au7–C2H2 | −4.40 | −5.51 | 4.79 | 4.69 |
| Au8–C2H2 | −3.38 | −6.24 | 5.81 | 5.42 |
| Au9–C2H2 | −4.29 | −5.34 | 4.91 | 4.52 |
| Au10–C2H2 | −3.71 | −5.86 | 5.48 | 5.05 |
In Fig. 2, the configuration of Au3,5,7,10–C2H2–HCl complexes is planar, whereas that of Au4,6,8,9–C2H2–HCl complexes is in the form of stereo structures. The adsorption energies of HCl on Aun–C2H2 (n = 4, 6, 8, 9) complexes approximate each other. The catalytic mechanism of acetylene hydrochlorination involves facilitating the dissociation of the H–Cl and C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C bonds. Therefore, the catalyst's ability to weaken the strong H–Cl and CC bonds and the degree of this weakening are crucial factors that affect its catalytic activity towards acetylene hydrochlorination. The bond lengths of HCl and C2H2 are all effectively lengthened in the Aun–C2H2–HCl complexes. The bond length increase (%) of RCC (C2H2) and RH–Cl (HCl) in Aun–C2H2–HCl complexes are listed in Table 2. In Fig. 3, all the studied Aun clusters have varying degrees of activation toward C2H2 and HCl. The weakening degree of HCl is generally stronger than that of C2H2 in Aun–C2H2–HCl complexes. The bond length of HCl in Au5–HCl is the most obvious and that of Au6,8–HCl are not obvious. In Fig. S3,† we found that the values of Au6 and Au8 coordinate points locate at peak. According to Fig. 3 and S3,† we can conclude that acetylene adsorption is better on gold clusters than on hydrogen chloride, and both bond lengthening upon adsorption and HOMO–LUMO gaps consistently indicate the highest activation for Au5 and the lowest one for Au6 and Au8.
C bonds. Therefore, the catalyst's ability to weaken the strong H–Cl and CC bonds and the degree of this weakening are crucial factors that affect its catalytic activity towards acetylene hydrochlorination. The bond lengths of HCl and C2H2 are all effectively lengthened in the Aun–C2H2–HCl complexes. The bond length increase (%) of RCC (C2H2) and RH–Cl (HCl) in Aun–C2H2–HCl complexes are listed in Table 2. In Fig. 3, all the studied Aun clusters have varying degrees of activation toward C2H2 and HCl. The weakening degree of HCl is generally stronger than that of C2H2 in Aun–C2H2–HCl complexes. The bond length of HCl in Au5–HCl is the most obvious and that of Au6,8–HCl are not obvious. In Fig. S3,† we found that the values of Au6 and Au8 coordinate points locate at peak. According to Fig. 3 and S3,† we can conclude that acetylene adsorption is better on gold clusters than on hydrogen chloride, and both bond lengthening upon adsorption and HOMO–LUMO gaps consistently indicate the highest activation for Au5 and the lowest one for Au6 and Au8.
 | ||
| Fig. 2 The optimal co-adsorption configurations and adsorption energies of HCl on different Aun–C2H2 complexes (The bond length in Å and energies in kcal mol−1). | ||
C (C2H2) and RH–Cl (HCl) in Aun–C2H2–HCl complexes
| Au3 | Au4 | Au5 | Au6 | Au7 | Au8 | Au9 | Au10 | |
|---|---|---|---|---|---|---|---|---|
| RCC |
2.42 | 2.56 | 1.99 | 1.03 | 1.54 | 1.22 | 1.23 | 1.32 |
| RH–Cl | 3.29 | 2.67 | 3.61 | 1.02 | 2.55 | 0.96 | 1.40 | 2.71 |
 | ||
| Fig. 3 The variation of bond length increase (%) of RCC (C2H2) and RH–Cl (HCl) in Aun–C2H2–HCl complexes. | ||
3.2 Mechanisms of acetylene hydrochlorination catalyzed by neutral Aun (n = 3–10) clusters
In the size range of Au3 to Au10, all gold clusters undergo the same catalytic cycle, as shown in Fig. 4. The whole process of acetylene hydrochlorination in Aun (n = 3–10) clusters does not vary across all clusters. However, the catalytic performances are different because of different sizes and active sites of the gold clusters. In Fig. 5, the difference in the energies of transition states and intermediates is evident. The path diagram shows two energy barriers, i.e., for the dissociation of hydrogen chloride and the transfer of proton from the Au cluster to chloroethenyl. The energies of acetylene adducts of the Au6, Au8, Au9 clusters are extremely low such that the second energy barrier is apparently higher than those of the others. We conjectured that this observation may be connected with their special structures (Fig. 6). If HCl is absorbed in the special triangle void, the proton of HCl would have difficulty in migrating out of the space. | ||
| Fig. 4 The catalytic cycle of acetylene hydrochlorination catalyzed by gold clusters. | ||
 | ||
| Fig. 5 The energy diagrams of the most favorable pathways of acetylene hydrochlorination on Aun (n = 3–10) clusters: reactants (Re), co-adsorbed reactants (co-ads), transition states (Ts), intermediate (Im), desorption products (De-Pr) and products (Pr). The data are zero-point-energy (ZPE) in kcal mol−1. | ||
 | ||
| Fig. 6 The special block of void comprised of three Au atoms in Au6,8,9–C2H2 and corresponding HOMO distributions. | ||
It is worthy of note that, by comparing the HOMO of Au6,8,9–C2H2 in Fig. 6, we found that the HOMO distributions were round the special block of void comprised of three Au atoms. But the HOMO of Au9–C2H2 seems a little different from the other two cases. We have studied the “odd−even” effect of the gold cluster (Fig. S3, in ESI†), which the even- and odd-numbered clusters are different in terms of their electronic performances. So we think that the difference of HOMO between Au9–C2H2 and Au6,8–C2H2 may be related to the “odd−even” effect of the gold cluster. After all, the “odd−even” effect is ubiquitous and plays a role in Au3–10 clusters.
 | ||
| Fig. 7 Energy diagrams of the most favorable pathways of acetylene hydrochlorination on Au3,5,7,10 clusters: reactants (R), co-adsorbed reactants (co-ads), transition states (Ts), intermediate (Im), desorption products (De-Pr) and products (Pr). The data are zero-point-energy (ZPE) in kcal mol−1. | ||
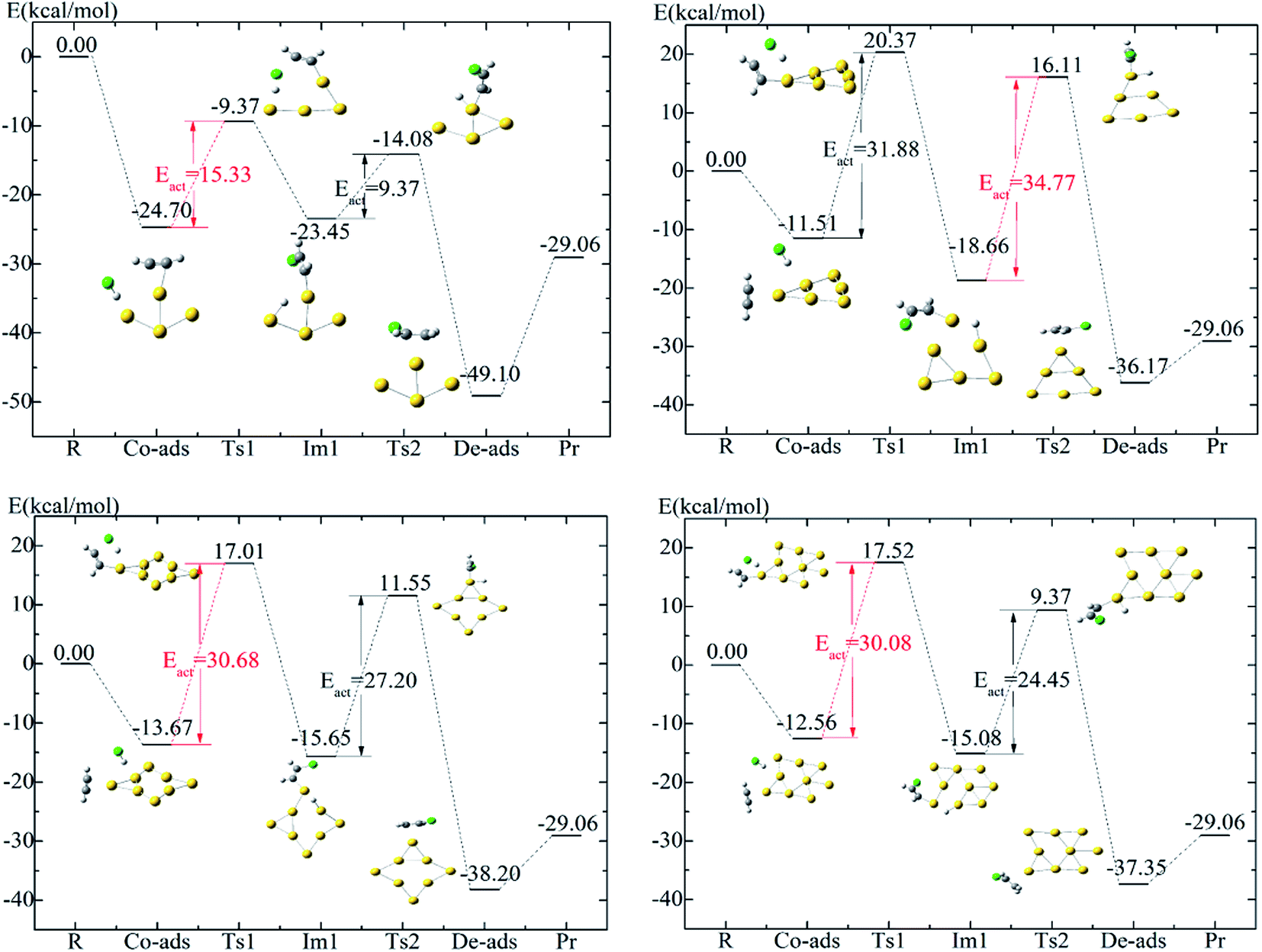 | ||
| Fig. 8 Energy diagrams of the most favorable pathways of acetylene hydrochlorination on Au4,6,8,9 clusters: reactants (R), co-adsorbed reactants (co-ads), transition states (Ts), intermediate (Im), desorption products (De-Pr) and products (Pr). The data are zero-point-energy (ZPE) in kcal mol−1. | ||
 | ||
| Fig. 9 The correlation between energy gaps and activation energies. The black line denotes energy gaps of HOMO–LUMO (Aun–C2H2 → HCl); the red line denotes the activation energy in the catalytic cycle of acetylene hydrochlorination. | ||
In conclusion, there is a direct relationship between the energy gaps of HOMO–LUMO (Aun–C2H2 → HCl) and activation energies indeed. However Au4 and Au9 don't follow the “odd−even” rule, those may be concerned with their special structures, and thus also support our conclusion in return. That is, we believe that apart from the size of gold clusters, different types of active sites or configurations can affect the catalytic activity performance.
In the end, by comparing with reference of J. Hutchings et al.,15 we found that this reaction could be achieved by forming active reaction species C2H2–AuCl3–HCl, which is similar to C2H2–Aun–HCl. Besides, they also concluded that the dissociation of hydrogen chloride was the rate-controlling step, and the activation energy barrier was 23.5 kcal mol−1. That indicates the gold-base catalysts are consistent in mechanism of acetylene hydrochlorination generally, but the catalytic performance has certain difference. By improving process of preparing and ultra-fine gold clusters of low atomicity, the acetylene hydrochlorination can be achieved in a higher efficiency with ultra-low trace amounts of Au catalyst.
4. Conclusion
Two different types of active sites exist in the Au clusters, namely, the top and bridge sites. In the Au4,6,8,9 clusters, HCl and C2H2 are activated by the top sites, whereas activation in the Au3,5,7,10 clusters occurs in bridge sites. In the co-adsorption process, Aun–C2H2 complexes are electron donors and HCl is an electron acceptor, which indicates that electrons transfer from the HOMO of Aun–C2H2 complexes (almost from Aun clusters) to the LUMO of HCl. Thus, the gold cluster functions as a bridge for electron transfer.The whole process of the acetylene hydrochlorination in the gold clusters involves two transition states (Ts1 and Ts2) and one intermediate (Im1). The process of Ts1 denotes the dissociation of HCl, whereas Ts2 refers to the proton transfer from the Au cluster to chloroethenyl. On the whole, the small-sized gold clusters result in a higher catalytic performance than large-sized clusters, and odd-number atom clusters perform better than even-number atom clusters. There is a direct relationship between the energy gaps of HOMO–LUMO (Aun–C2H2 → HCl) and activation energies indeed. We believe that apart from the size of gold clusters, different types of active sites or configurations can affect the catalytic activity performance.
Acknowledgements
We gratefully acknowledge the financial supports for the Special Funds for Major State Basic Research Program of China (no. 2012CB720302), the National Natural Science Fundation of China (NSFC, Grant no. 21363020) and the Ph.D. Programs Foundation of the Xinjiang Production and Construction Corps (no. 2013BB010).References and notes
- G. J. Hutchings, Catal. Today, 2005, 100, 55–61 CrossRef CAS PubMed.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896–7936 CrossRef PubMed.
- Z. Li, C. Brouwer and C. He, Chem. Rev., 2008, 108, 3239–3265 CrossRef CAS PubMed.
- R. Sardar, A. M. Funston, P. Mulvaney and R. W. Murray, Langmuir, 2009, 25, 13840–13851 CrossRef CAS PubMed.
- C. H. Christensen and J. K. Norskov, Science, 2010, 327, 278–279 CrossRef PubMed.
- A. Corma, A. Leyva-Pérez and M. J. Sabater, Chem. Rev., 2011, 111, 1657–1712 CrossRef CAS PubMed.
- G. J. Hutchings, J. Catal., 1985, 96, 292–295 CrossRef CAS.
- M. Haruta, N. Yamada, T. Kobayashi and S. Iijima, J. Catal., 1989, 115, 301–309 CrossRef CAS.
- G. J. Hutchings, Gold Bull., 1996, 29, 123–130 CrossRef CAS.
- J. E. Bailie and G. J. Hutchings, Chem. Commun., 1999, 2151–2152 RSC.
- P. Landon, P. J. Collier, A. J. Papworth, C. J. Kiely and G. J. Hutchings, Chem. Commun., 2002, 2058–2059 RSC.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–939 CrossRef CAS PubMed.
- M. D. Hughes, Y. Xu, P. Jenkins, P. McMorn, P. Landon, D. I. Enache, A. F. Carley, G. A. Attard, G. J. Hutchings, F. King, E. H. Stitt, P. Johnston, K. Griffin and C. J. Kiely, Nature, 2005, 437, 1132–1135 CrossRef CAS PubMed.
- F. Shi, X. Li, Y. Xia, L. Zhang and Z. Yu, J. Am. Chem. Soc., 2007, 129, 15503–15512 CrossRef CAS PubMed.
- M. Conte, A. F. Carley, C. Heirene, D. J. Willock, P. Johnston, A. A. Herzing, C. J. Kiely and G. J. Hutchings, J. Catal., 2007, 250, 231–239 CrossRef CAS PubMed.
- B. Yang, R. Burch, C. Hardacre, G. Headdock and P. Hu, ACS Catal., 2012, 2, 1027–1032 CrossRef CAS.
- H. Y. Kim, H. M. Lee and G. Henkelman, J. Am. Chem. Soc., 2012, 134, 1560–1570 CrossRef CAS PubMed.
- J. Lu, C. Aydin, N. D. Browning and B. C. Gates, Angew. Chem., 2012, 124, 5944–5948 CrossRef PubMed.
- C. Liu, Y. Tan, S. Lin, H. Li, X. Wu, L. Li, Y. Pei and X. Zeng, J. Am. Chem. Soc., 2013, 135, 2583–2595 CrossRef CAS PubMed.
- A. P. Woodham, G. Meijer and A. Fielicke, J. Am. Chem. Soc., 2013, 135, 1727–1730 CrossRef CAS PubMed.
- A. Corma, P. Concepcion, M. Boronat, M. J. Sabater, J. Navas, M. J. Yacaman, E. Larios, A. Posadsas, M. A. Lopez-Quintela, D. Buceta, E. Mendoza, G. Guilera and A. Mayoral, Nat. Chem., 2013, 5, 775–781 CrossRef CAS PubMed.
- W. Wang, J. Gu, W. Hua, X. Jia and K. Xi, Chem. Commun., 2014, 50, 8889–8891 RSC.
- B. Nkosi, N. J. Coville and G. J. Hutchings, J. Chem. Soc., Chem. Commun., 1988, 71–72 RSC.
- M. Zhu, L. Kang, Y. Su, S. Zhang and B. Dai, Can. J. Chem., 2013, 91, 120–125 CrossRef CAS.
- J. Zhang, Z. He, W. Li and Y. Han, RSC Adv., 2012, 2, 4814–4821 RSC.
- C. Huang, M. Zhu, L. Kang and B. Dai, Catal. Commun., 2014, 54, 61–65 CrossRef CAS PubMed.
- C. Huang, M. Zhu, L. Kang, X. Li and B. Dai, Chem. Eng. J., 2014, 242, 69–75 CrossRef CAS PubMed.
- X. Li, M. Zhu and B. Dai, Appl. Catal., B, 2013, 142–143, 234–240 CrossRef CAS PubMed.
- X. Li, Y. Wang, L. Kang, M. Zhu and B. Dai, J. Catal., 2014, 311, 288–294 CrossRef CAS PubMed.
- X. Li, X. Pan, L. Yu, P. Ren, X. Wu, L. Sun, F. Jiao and X. Bao, Nat. Commun., 2014 DOI:10.1038/ncomms4688.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, Ö. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Gaussian Inc., Wallingford CT, 2010 Search PubMed.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- M. Andersson and P. Uvdal, J. Phys. Chem. A, 2005, 109, 2937–2941 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem. C, 1989, 90, 2154–2162 CrossRef CAS PubMed.
- C. Gonzalez and H. B. Schlegel, J. Phys. Chem. C, 1990, 94, 5523–5527 CrossRef CAS.
- F. B. Vanduijneveldt, J. G. C. M. V. vande Rijdt and J. H. Lenthe, Chem. Rev., 1994, 94, 1873–1885 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05463f |
| This journal is © The Royal Society of Chemistry 2014 |