
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metal-organic frameworks (MOFs) as precursors towards TiOx/C composites for photodegradation of organic dye†
Zhengang
Guo
,
Jin Kiat
Cheng
,
Zhigang
Hu
,
Mei
Zhang
,
Qing
Xu
,
Zixi
Kang
and
Dan
Zhao
 *
*
Department of Chemical and Biomolecular Engineering, National University of Singapore, Singapore 117585. E-mail: chezhao@nus.edu.sg
First published on 7th August 2014
Abstract
A series of TiOx/C composites were prepared by pyrolyzing a Ti-containing MOF MIL-125, which exhibited excellent catalytic activities towards the photodegradation of methylene blue (MB) in aqueous solution. The best photocatalytic performance was achieved in MIL-125 pyrolyzed at 1000 °C due to its high surface area, reduced Ti3O5 composition, and conductive carbon matrix. This work represents a novel approach towards TiOx/C composites as photocatalysts.
The harvest of solar energy has become an important procedure in the research of clean energy and environmental sustainability, such as water splitting,1 degradation of organic pollutants,2 and photocatalytic reduction of CO2.3 One of the bottle-necks in this procedure is the development of photocatalysts that can make full use of the whole solar spectrum with high energy conversion efficiency. Metal oxides are an important family of photocatalysts, among which TiO2 is the widest studied one due to its semiconductor property, high natural abundance, and low toxicity.4 In the last decade, a lot of research has been done to improve the photocatalytic activity of TiO2, such as morphology control,5 crystal facet engineering,6 doping with other elements,7,8etc., with the aim for better absorption of solar energy and smaller chance of electron–hole recombination. Recently, TiO2/C composites have attracted lots of attention because of (1) higher surface area provided by the porous carbon matrix with better adsorption of reagents; (2) extended light absorption range and higher absorption intensity; and (3) longer electron–hole recombination time, all of which are beneficial to the photocatalytic activities.9,10 The current methods for the preparation of TiO2/C composites include simple mixing, thermal oxidation, sol–gel, hydrothermal, deposition, etc.10 Novel approaches are needed to have better control and fine tuning of the composition, texture, and polymorphs of TiO2/C composites.
Metal-organic frameworks (MOFs, aka porous coordination polymers, PCPs) are hybrid crystalline materials composed of metal nodes and ligand spacers.11–15 Because of their versatile chemical composition, high porosity, uniform yet tunable pore size and shape, they have found wide applications in storage,16 separation,17 catalysis,18,19 sensing,20,21 biomedical fields,22etc. Recently, MOFs have been used either as templates or as sacrificial precursors in preparing porous carbon,23–25 metal oxides nanoparticles,26,27 and metal oxide/carbon composite materials,28,29 with the major applications in supercapacitors, batteries, and as electrocatalysts in fuel cells for energy storage and conversion.
Ti-containing MOFs have been reported in the literature.30–33 Their photocatalytic activities have been demonstrated through CO2 reduction,31 water splitting,34 and degradation of organic dye.33 However, because of the labile coordination bonds in MOFs,35,36 using Ti-containing MOFs directly as photocatalysts has the risk of material decomposition, especially in aqueous environments where most of the photocatalytic reactions are carried out. Alternatively, Ti-containing MOFs can be pyrolyzed into TiO2/C composites with much higher stabilities. A previous work has been reported on pyrolyzing Ti-modified IRMOF-3.37 However, due to the complicated postsynthetic modification of MOF precursor with titanium isopropoxide, the Ti content in the final pyrolyzed product is only 4.3 wt% which greatly limits its photocatalytic activity. In this communication, we report the facile synthesis of robust TiOx/C composites using Ti-containing MOFs as the sacrificial precursors. Thanks to the hybrid and crystalline structure of the MOF precursors, the chemical composition and pore texture of TiOx/C composites can be readily adjusted, rending a series of photocatalysts which exhibit excellent photocatalytic activities towards the degradation of methylene blue.
MIL-125 is one of the earliest Ti-containing MOFs reported so far.30 It is composed of 1,4-benzenedicarboxylate (BDC) bridged by Ti/O clusters as the secondary building units (SBUs). In this study, MIL-125 was selected as the MOF precursor, which was pyrolyzed under Ar atmosphere at temperatures of 400, 600, 800, and 1000 °C, respectively, with the products named as T4, T6, T8, and T10 (see ESI† for experimental details).
The powder X-ray diffraction (PXRD) pattern of synthesized MIL-125 matches well with the one simulated from published single crystal structure (Fig. 1). The white powder of MIL-125 turned into blackish after pyrolysis, indicating the formation of carbonaceous materials. The crystallinity of MIL-125 was completely lost in T4, and the PXRD peak at 25.2° belonging to (101) plane of anatase TiO2 (JCPDS card 21-1272) can be easily identified, revealing the decomposition of crystalline MIL-125 framework and evolution of TiO2/C composites under this temperature (400 °C, Fig. 1). For the sample of T6 which was pyrolyzed at a high temperature (600 °C), more peaks representing anatase have emerged, indicating better crystallinity of anatase phase. Increasing the pyrolysis temperature to 800 °C trigged the phase transition of anatase to rutile, rending new peaks at 27.3, 36.0, and 41.2° belonging to (110), (101), and (111) planes of rutile TiO2 (JCPDS card 21-1276) in T8, respectively. This phase transition is anticipated as anatase is a metastable polymorph of TiO2 which can be converted to a more stable rutile polymorph at higher temperatures.38 Neither anatase nor rutile polymorph can be found in T10 which was pyrolyzed at an even higher temperature (1000 °C). Instead, the newly emerged peaks match well with the PXRD pattern of γ-Ti3O5 (JCPDS card 40-0806),39 whose formation can be attributed to the carbothermal reduction of TiO2 under this condition.40,41
 | ||
| Fig. 1 PXRD spectra of samples and reference. | ||
Scanning electron microscope (SEM) images of pyrolyzed MIL-125 indicate a variable distribution of particles of about 0.1–1 μm in size and clusters of particles packed together (Fig. S1†). Even distributions of C, O, and Ti in pyrolyzed samples can be confirmed by the EDS elemental mapping (Fig. S2†). Transmission electron microscope (TEM) images of T4 reveal the structure of carbonaceous matrix imbedded with crystalline nanoparticles with the size of around 5 nm, which increases to around 10 nm in T6 with a lattice spacing of d = 0.352 nm representing the (101) plane of anatase polymorph (Fig. 2). The evolution of rutile polymorph in T8 was confirmed by the lattice spacing of 0.325 nm indicating the (110) plane of rutile. Larger particles with the size of 20–50 nm can be found in T10, whose lattice spacing of 0.292 nm matches well with the (![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 12) plane of Ti3O5.
12) plane of Ti3O5.
 | ||
| Fig. 2 TEM images of (a) T4, (b) T6, (c) T8, and (d) T10. | ||
The chemical state changes of C and Ti elements in MIL-125 pyrolyzed at different temperatures were traced through X-ray photoelectron spectroscopy (XPS). As can be seen from the C 1s XPS spectra (Fig. 3a), the shake-up peak at around 291 eV coming from pi–pi* transition becomes more distinct for samples pyrolyzed at higher temperatures, indicating a higher degree of graphitization in these samples. Ti mainly exists as Ti4+ (TiO2) in samples being treated at lower temperatures (T4 and T6), while higher portions of Ti3+ and Ti2+ can be found in samples obtained at higher temperatures (T8 and T10, Fig. 3b). This conclusion agrees well with the PXRD and TEM data in supporting the formation of reduced TiO2 (such as Ti3O5) in MIL-125 pyrolyzed at higher temperatures.
 | ||
| Fig. 3 XPS spectra of TiOx/C composites: (a) C 1s; (b) Ti 2p. | ||
Thermogravimetric analyses (TGA) of the pyrolyzed samples were carried out under an air flow to burn off the carbon matrix and yield white TiO2 powder as the final products (Fig. S3†).37 The highest Ti content was observed in T10 (50.2 wt%), followed by T6 (40.7 wt%), T8 (38.6 wt%), and T4 (37.0 wt%). The Ti contents in our samples are much higher than the previous report of 4.3 wt%37 reinforcing the attractiveness of preparing TiOx/C composites using Ti-containing MOFs directly as precursors. It is interesting to note that the TGA curve of T10 exhibits an upward curve at temperatures below 400 °C. This can be attributed to the oxidation of Ti3O5 into TiO2 because the calculated weight increase (5.6 wt%) assuming this process matches well with the measured weight increase (5.3 wt%).42 Thermal stability of the pyrolyzed samples is also demonstrated as combustion in air won't happen until the temperature is higher than 400 °C, which grants the applications of these materials in conditions up to 400 °C.
The surface area and pore size distribution data were obtained by analyzing N2 sorption isotherms collected at 77 K (Fig. 4). The isotherm of repeated MIL-125 exhibits a Type I shape indicating the microporous structure which reflects the crystal model and matches well with the literature.30 A slight hysteresis between adsorption and desorption branches was found in MIL-125, which can be attributed to the mesoporous voids formed among crystalline particles. The surface area of MIL-125 was calculated to be 1321 m2 g−1 using BET model (1563 m2 g−1 assuming Langmuir model). All the pyrolyzed samples, however, exhibit Type IV isotherms with much smaller surface areas (Table 1), possibly due to the loss of crystallinity and crack of framework during pyrolysis. In T4 and T6, the pore size still remains within microporous range (<2 nm). Higher pyrolysis temperatures help to increase the surface area and trigger the formation of both smaller pores (∼0.8 nm) and larger pores (>3 nm) in T8 and T10. It is clearly demonstrated herein that the surface area and pore size of TiOx/C composites can be readily changed through pyrolysis temperature using Ti-containing MOFs as precursors. Although the surface areas of our samples are merely moderate, the versatile synthetic approaches of using MOFs as precursors to prepare carbon materials, such as including supplementary carbon sources within MOF cavities,24 give enormous opportunities for the fine-tune of porous texture that is rarely seen in other synthetic methods.
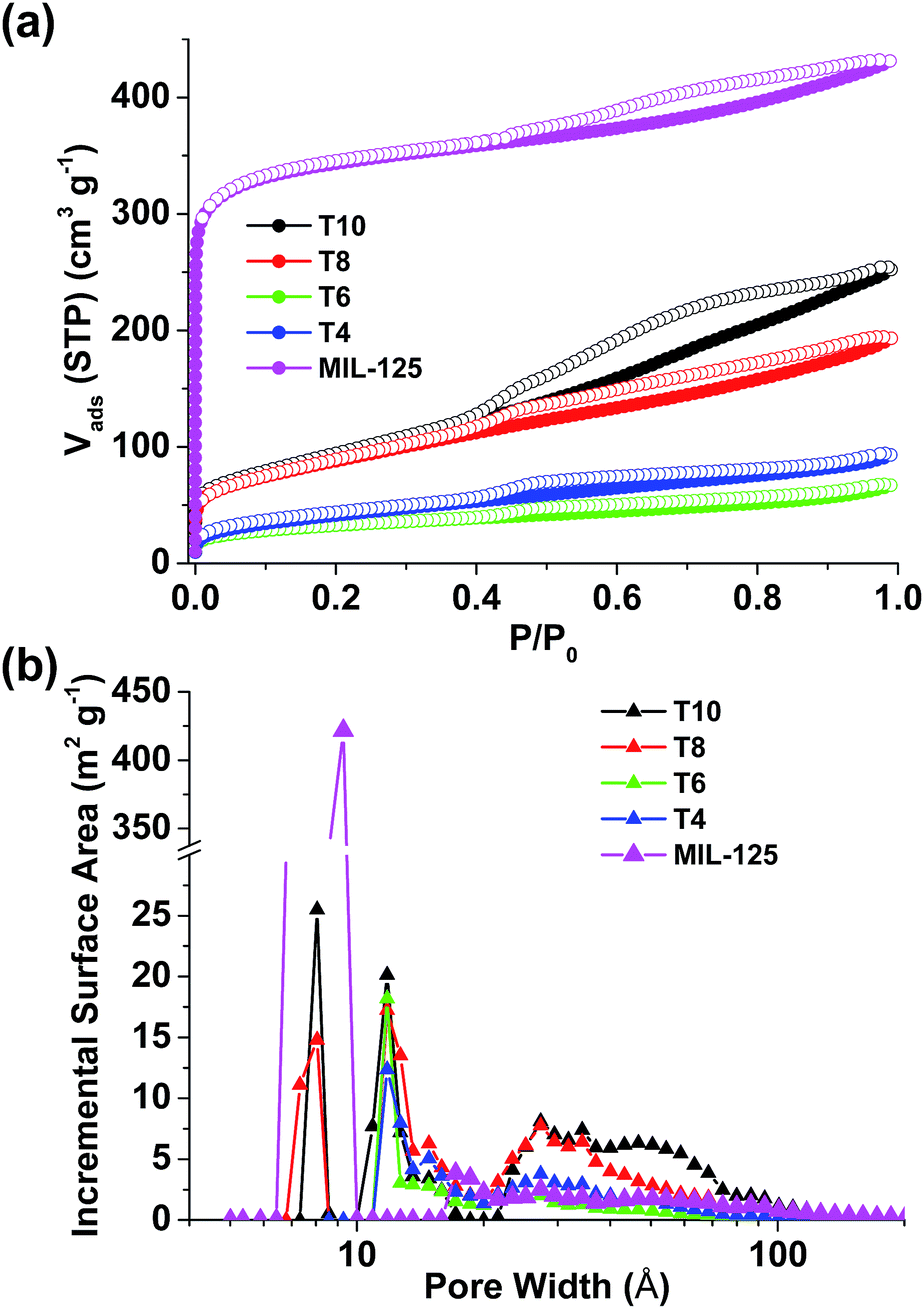 | ||
| Fig. 4 (a) N2 sorption isotherms and (b) pore size distribution of MIL-125 and pyrolyzed samples. | ||
The photocatalytic activities of our samples were measured through photodegradation of methylene blue (MB), a common dye used for the simulation of pollutant in industrial effluent streams (see ESI† for experimental details). Two extra samples were added as comparisons. One is P25 TiO2 that is commercially available with mixed anatase/rutile crystallites commonly used as the benchmark photocatalyst. The other one is T10 washed with HF (T10/HF) to remove Ti3O5 affording porous carbon. Before subject to irradiation, all the sample solutions were left under stirring in dark for 1 hour to reach physisorption equilibrium of MB (Fig. S4a†). More MB up to 25% can be physically adsorbed into the samples with a higher surface area such as T10, while there is barely any physisorption of MB observed in P25 due to its low surface area (Fig. S4b†).
All the pyrolyzed samples exhibited photocatalytic activities towards the degradation of MB (Fig. S5†). After 60 minutes of irradiation, 38.8% of MB was degraded under the catalysis of T10, while only 32.9% of MB was degraded with P25 as the catalyst (Fig. 5a and Table 1). The photodegradation data at irradiation time ≤ 20 minutes were used to fit the kinetics as first order model (Fig. 5b). The apparent first-order rate constant k was obtained that can reflect the speed of photodegradation. T10 has the largest k, which is 32% higher than that of P25. It can be safely concluded that compared to the benchmark P25 TiO2, at least one pyrolyzed sample (T10) reported herein has a better photocatalytic activity towards the photodegradation of methylene blue. The poorest activity was observed in T10/HF, where most of the Ti3O5 has been removed through HF wash. This fact confirms that the superior photocatalytic activity of T10 should come from the imbedded Ti3O5 nanoparticles.
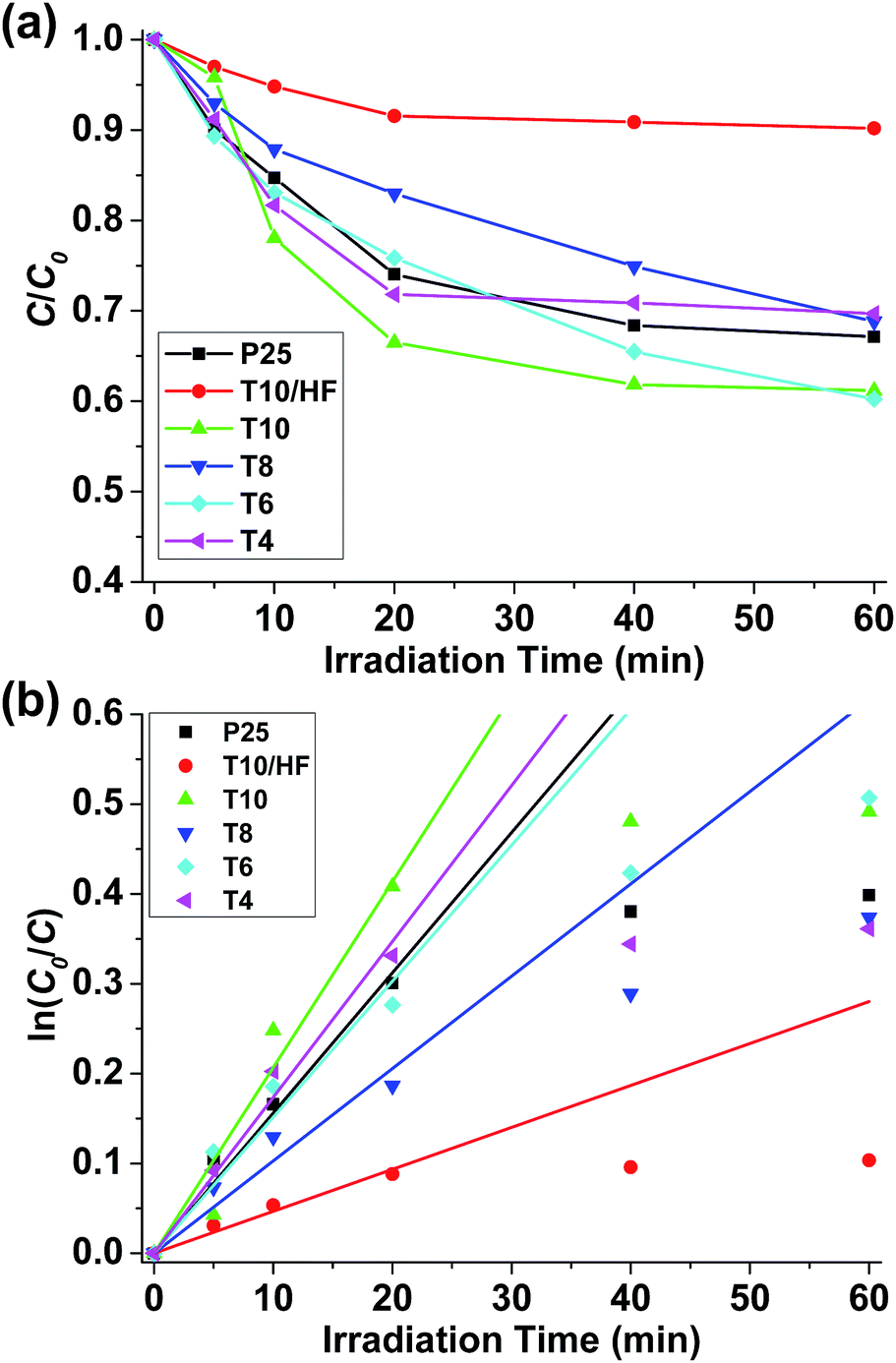 | ||
| Fig. 5 (a) Concentration changes of MB under irradiation with various catalysts. (b) Fitting of kinetic data using first order model. | ||
Several factors need to be considered in order to enunciate the relationship between the structure of pyrolyzed samples and their photocatalytic activities. The most obvious factor is the content of TiOx as they serve as the catalytic sites. T10 has the highest Ti content among the pyrolyzed samples which may justify its good activity. However, its better activity compared to pure TiO2 needs further explanation. Since the photodegradation described herein is heterogeneously catalyzed, the access of substrates towards active sites is another important factor. Compared to pure TiO2, pyrolyzed samples have TiOx as nanoparticles spread apart and supported on porous carbon matrix that make it easier for the substrate (MB in this case) to access and react. This point is commonly accepted in heterogeneous catalysis43 and is supported by the high surface area of T10. However, surface area should not be the sole factor in determining photocatalytic activities. The surface areas of T8 and T10 are similar while T8 only demonstrates sluggish reaction kinetics. A closer observation of material structure reveals a huge difference of TiOx between T8 and T10. Ti exists as TiO2 in T8, with a substantial portion of rutile polymorph that is catalytically less active than its anatase analogue.44 Although it has been reported that TiO2 with anatase/rutile mixed phases has better photocatalytic activities due to efficient electron–hole separation,45,46 such enhanced catalytic activity was not observed in T8 possibly due to the separation of anatase and rutile nanoparticles in carbon matrix that prevents close contact and impairs synergetic effect. In T10, nevertheless, TiO2 is carbothermally reduced into Ti3O5 with higher density of oxygen vacancies for better trapping of electrons affording more efficient photocatalysis.47 Although Ti3O5 has been extensively studied for its phase transition behaviour,48–50 there is seldom any report for its photocatalytic performance51 and this work is among the initial studies to the best of our knowledge. The last factor that needs to be taken into consideration is the conductivity of carbon matrix. One of the properties of TiOx/C composites is the electron conductivity provided by the carbon matrix which can afford longer electron–hole recombination time for more efficient catalysis. From this perspective, samples pyrolyzed at higher temperatures (such as T10) should have better activities because of the higher electron conductivity brought by higher degree of graphitization proved by XPS previously.
In summary, a series of TiOx/C composites were prepared through pyrolysis of a Ti-containing MOF MIL-125. They exhibited considerable photocatalytic activities towards the photodegradation of methylene blue. Among these materials, T10 has the best activity due to its high surface area, reduced Ti3O5 composition, and conductive carbon support. This work demonstrates the potency of preparing TiOx/C composites as photocatalysts using Ti-containing MOFs as precursors. More work will be focused on fine-tuning the porosity, polymorph, and elemental doping of TiOx/C composites through the judicious selection of functional MOF precursors and pyrolysis conditions.
Acknowledgements
This work is supported by National University of Singapore (NUS Start-up Funding R-279-000-369-133) and Singapore Ministry of Education (MOE AcRF Tier 1 R-279-000-410-112, R-279-000-361-731). We acknowledge Professor Gade Pandu Rangaiah and Chen Xu for their assistance in photodegradation experiments.Notes and references
- X. B. Chen, S. H. Shen, L. J. Guo and S. S. Mao, Chem. Rev., 2010, 110, 6503–6570 CrossRef CAS PubMed.
- S. Ahmed, Crit. Rev. Environ. Sci. Technol., 2012, 42, 601–675 CrossRef CAS.
- S. N. Habisreutinger, L. Schmidt-Mende and J. K. Stolarczyk, Angew. Chem., Int. Ed., 2013, 52, 7372–7408 CrossRef CAS PubMed.
- A. Fujishima, X. T. Zhang and D. A. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS PubMed.
- X. B. Chen and S. S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS PubMed.
- W. J. Ong, L. L. Tan, S. P. Chai, S. T. Yong and A. R. Mohamed, ChemSusChem, 2014, 7, 690–719 CrossRef CAS PubMed.
- R. Asahi, T. Morikawa, T. Ohwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS PubMed.
- P. Xu, T. Xu, J. Lu, S. M. Gao, N. S. Hosmane, B. B. Huang, Y. Dai and Y. B. Wang, Energy Environ. Sci., 2010, 3, 1128–1134 CAS.
- K. Woan, G. Pyrgiotakis and W. Sigmund, Adv. Mater., 2009, 21, 2233–2239 CrossRef CAS.
- R. Leary and A. Westwood, Carbon, 2011, 49, 741–772 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- G. Férey, Chem. Soc. Rev., 2008, 37, 191–214 RSC.
- J. R. Long and O. M. Yaghi, Chem. Soc. Rev., 2009, 38, 1213–1214 RSC.
- H. C. Zhou, J. R. Long and O. M. Yaghi, Chem. Rev., 2012, 112, 673–674 CrossRef CAS PubMed.
- D. Zhao, D. Q. Yuan and H. C. Zhou, Energy Environ. Sci., 2008, 1, 222–235 CAS.
- J. R. Li, J. Sculley and H. C. Zhou, Chem. Rev., 2012, 112, 869–932 CrossRef CAS PubMed.
- Z. Wang, G. Chen and K. L. Ding, Chem. Rev., 2009, 109, 322–359 CrossRef CAS PubMed.
- M. A. Nasalevich, M. van der Veen, F. Kapteijn and J. Gascon, CrystEngComm, 2014, 16, 4919–4926 RSC.
- L. E. Kreno, K. Leong, O. K. Farha, M. Allendorf, R. P. Van Duyne and J. T. Hupp, Chem. Rev., 2012, 112, 1105–1125 CrossRef CAS PubMed.
- M. Zhang, G. X. Feng, Z. G. Song, Y. P. Zhou, H. Y. Chao, D. Q. Yuan, T. T. Y. Tan, Z. G. Guo, Z. G. Hu, B. Z. Tang, B. Liu and D. Zhao, J. Am. Chem. Soc., 2014, 136, 7241–7244 CrossRef CAS PubMed.
- P. Horcajada, R. Gref, T. Baati, P. K. Allan, G. Maurin, P. Couvreur, G. Férey, R. E. Morris and C. Serre, Chem. Rev., 2012, 112, 1232–1268 CrossRef CAS PubMed.
- B. Liu, H. Shioyama, T. Akita and Q. Xu, J. Am. Chem. Soc., 2008, 130, 5390–5391 CrossRef CAS PubMed.
- H. L. Jiang, B. Liu, Y. Q. Lan, K. Kuratani, T. Akita, H. Shioyama, F. Q. Zong and Q. Xu, J. Am. Chem. Soc., 2011, 133, 11854–11857 CrossRef CAS PubMed.
- M. Hu, J. Reboul, S. Furukawa, N. L. Torad, Q. M. Ji, P. Srinivasu, K. Ariga, S. Kitagawa and Y. Yamauchi, J. Am. Chem. Soc., 2012, 134, 2864–2867 CrossRef CAS PubMed.
- J. Zhao, F. Q. Wang, P. P. Su, M. R. Li, J. Chen, Q. H. Yang and C. Li, J. Mater. Chem., 2012, 22, 13328–13333 RSC.
- M. Y. Masoomi and A. Morsali, Coord. Chem. Rev., 2012, 256, 2921–2943 CrossRef CAS PubMed.
- D. Zhao, J. L. Shui, C. Chen, X. Q. Chen, B. M. Reprogle, D. P. Wang and D. J. Liu, Chem. Sci., 2012, 3, 3200–3205 RSC.
- D. Zhao, J. L. Shui, L. Grabstanowicz, C. Chen, S. Commet, T. Xu, J. Lu and D. J. Liu, Adv. Mater., 2014, 26, 1093–1097 CrossRef CAS PubMed.
- M. Dan-Hardi, C. Serre, T. Frot, L. Rozes, G. Maurin, C. Sanchez and G. Ferey, J. Am. Chem. Soc., 2009, 131, 10857–10859 CrossRef CAS PubMed.
- Y. H. Fu, D. R. Sun, Y. J. Chen, R. K. Huang, Z. X. Ding, X. Z. Fu and Z. H. Li, Angew. Chem., Int. Ed., 2012, 51, 3364–3367 CrossRef CAS PubMed.
- C. H. Hendon, D. Tiana, M. Fontecave, C. Sanchez, L. D'arras, C. Sassoye, L. Rozes, C. Mellot-Draznieks and A. Walsh, J. Am. Chem. Soc., 2013, 135, 10942–10945 CrossRef CAS PubMed.
- J. K. Gao, J. W. Miao, P. Z. Li, W. Y. Teng, L. Yang, Y. L. Zhao, B. Liu and Q. C. Zhang, Chem. Commun., 2014, 50, 3786–3788 RSC.
- Y. Horiuchi, T. Toyao, M. Saito, K. Mochizuki, M. Iwata, H. Higashimura, M. Anpo and M. Matsuoka, J. Phys. Chem. C, 2012, 116, 20848–20853 CAS.
- J. J. Low, A. I. Benin, P. Jakubczak, J. F. Abrahamian, S. A. Faheem and R. R. Willis, J. Am. Chem. Soc., 2009, 131, 15834–15842 CrossRef CAS PubMed.
- L. Bellarosa, J. M. Castillo, T. Vlugt, S. Calero and N. López, Chem.-Eur. J., 2012, 18, 12260–12266 CrossRef CAS PubMed.
- J. Kim, N. D. McNamara, T. H. Her and J. C. Hicks, ACS Appl. Mater. Interfaces, 2013, 5, 11479–11487 CAS.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- S. H. Hong, Acta Chem. Scand., Ser. A, 1982, 36, 207–217 CrossRef PubMed.
- G. A. Swift and R. Koc, J. Mater. Sci., 1999, 34, 3083–3093 CrossRef CAS.
- X. He, J. W. Ye, Y. Liu, B. Q. Chen, Z. T. Jiang, H. W. Zou, L. Deng and M. J. Tu, Adv. Powder Technol., 2010, 21, 448–451 CrossRef CAS PubMed.
- T. V. Charlu, O. J. Kleppa and T. B. Reed, J. Chem. Thermodyn., 1974, 6, 1065–1074 CrossRef CAS.
- M. A. Fox and M. T. Dulay, Chem. Rev., 1993, 93, 341–357 CrossRef CAS.
- A. Wold, Chem. Mat., 1993, 5, 280–283 CrossRef CAS.
- D. C. Hurum, A. G. Agrios, K. A. Gray, T. Rajh and M. C. Thurnauer, J. Phys. Chem. B, 2003, 107, 4545–4549 CrossRef CAS.
- D. O. Scanlon, C. W. Dunnill, J. Buckeridge, S. A. Shevlin, A. J. Logsdail, S. M. Woodley, C. R. A. Catlow, M. J. Powell, R. G. Palgrave, I. P. Parkin, G. W. Watson, T. W. Keal, P. Sherwood, A. Walsh and A. A. Sokol, Nat. Mater., 2013, 12, 798–801 CrossRef CAS PubMed.
- Y. Z. Li, D. S. Hwang, N. H. Lee and S. J. Kim, Chem. Phys. Lett., 2005, 404, 25–29 CrossRef CAS PubMed.
- C. N. R. Rao, S. Ramdas, R. E. Loehman and J. M. Honig, J. Solid State Chem., 1971, 3, 83–88 CrossRef CAS.
- M. Onoda, J. Solid State Chem., 1998, 136, 67–73 CrossRef CAS.
- S. Ohkoshi, Y. Tsunobuchi, T. Matsuda, K. Hashimoto, A. Namai, F. Hakoe and H. Tokoro, Nat. Chem., 2010, 2, 539–545 CrossRef CAS PubMed.
- N. Stem, E. F. Chinaglia and S. G. dos Santos Filho, ECS Trans., 2011, 39, 347–354 CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, SEM images, EDS elemental mapping, TGA, and UV-Vis spectra. See DOI: 10.1039/c4ra05429f |
| This journal is © The Royal Society of Chemistry 2014 |