
Organophosphonates as anchoring agents onto metal oxide-based materials: synthesis and applications
Remi Boissezon
,
Julien Muller,
Vincent Beaugeard,
Sophie Monge* and
Jean-Jacques Robin
Institut Charles Gerhardt Montpellier UMR5253 CNRS-UM2-ENSCM-UM1-Equipe Ingénierie et Architectures Macromoléculaires, Université Montpellier II, cc1702, Place Eugène Bataillon, 34095 Montpellier, France. E-mail: sophie.monge-darcos@univ-montp2.fr; Fax: +33–467144028; Tel: +33-467144158
First published on 30th July 2014
Abstract
Besides usual coupling agents such as, for instance, silane, thiol, or carboxylic acids, phosphonated-based low molecular weight molecules and polymers appear of great interest for the modification of metal oxides. In such contexts, this Review is focused on the use of phosphonated derivatives for the surface treatment of metal oxides as such compounds are known to establish strong and stable links with inorganic phases. Functionalization of metal oxides leads to the obtaining of hybrid materials that can find applications in numerous fields. As a result, an interesting prospect will be to enhance the development of new hybrid materials in the future, especially bio-based ones.
 Remi Boissezon, Vincent Beaugeard, Jean-Jacques Robin, Julien Muller and Sophie Monge | From left to right: Rémi Boissezon, Vincent Beaugeard, Prof. Jean-Jacques Robin, Julien Muller, and Dr Sophie Monge. Prof. Jean-Jacques Robin is currently leading the “Ingénierie et Architectures Macromoléculaires” laboratory (IAM) of the “Institut Charles Gerhardt de Montpellier” (ICGM). His area of expertise mainly deals with the synthesis of graft copolymers and the chemical recycling of polymers. Rémi Boissezon, Vincent Beaugeard, and Julien Muller are PhD students working at the IAM/ICGM on the functionalization of (in)organic materials, notably with phosphorus-based molecules. Dr Sophie Monge is assistant professor at the IAM/ICGM and her research interests mainly focus on the synthesis of well-defined (co)polymers with stimuli-responsive properties, and the synthesis of polymers bearing heteroatoms, especially phosphorus. |
1 Introduction
Materials combining inorganic and organic components are of great interest as they exhibit original and valuable properties.1 As a result, they are nowadays widely studied by numerous researchers and companies due to the demand for innovative materials providing appropriate responses for specific applications. In the past, engineers developed blends by mixing various components and observed the emergence of new properties combining those of the pure counterparts. Among those materials, organic polymers incorporating minerals or fibers are the most famous encountered examples and, in some cases, some synergistic effects could be reached and surprising properties were obtained. We can also mention the development of nanocomposites these last years, motivated by the trend to decrease the size of the phases to reach novel properties like better permeability to gases, thermal stability or viscosity leading to various applications such as in the biomedical field, for instance.2 At last, it must be pointed out that this concept of organic–inorganic materials is largely observed in nature where mineral part can grow from an organic skeleton giving complex morphologies, most often exhibiting surprising geometries and shapes.3In all cases of organic–inorganic materials, the main key point stands with the interaction existing between the two components. In terms of stability of the obtained morphologies, weak interactions can lead in some cases to changes in morphologies during ageing. Stronger interactions are generally preferred, most often through ionic or covalent bonding. On the reverse, too strong interactions can prevent processing since they can act as crosslinkers. Various groups are able to interact with inorganic surfaces such as metal or metal oxide. Beta-diketones, thiols, carboxylic acids, hydroxyls, alkoxysilanes, amines, amides, sulfonic acids, phosphines, phosphonic acids are the most often encountered ones.4–8 These functions can be inserted in (co)polymers synthesized from functional monomers or capped by various alkyl chains. Among the last cited compounds, phosphorus-based molecules are very valuable, mainly phosphonated derived compounds since they are easily prepared according to simple chemical routes, as shown in Scheme 1.
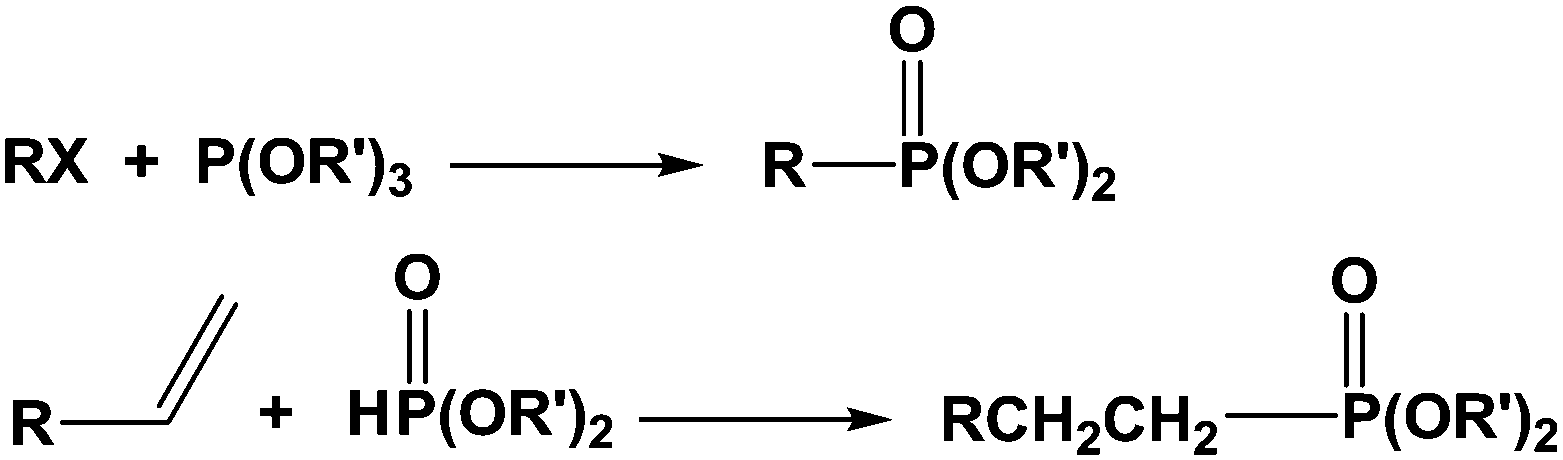 | ||
| Scheme 1 Main routes for the synthesis of phosphonated compounds. | ||
It must be pointed out that other ways have been explored for the synthesis of monomers bearing one or two phosphonate moieties and a polymerizable group. These ways have been reviewed by Chougrani and al.9 Numerous interesting chemical structures were prepared as resulting phosphonic-based compounds showed excellent complexing properties, biocompatibility, and chemical resistance.10 As a result, they were directly used for surface modification, or for copolymerization, thus providing (co)polymers able to strongly interact with metallic surfaces to generate (i) protective coatings or sealants,11 (ii) ionomer resins,12 or materials for flame retardancy,13 for instance.
Additionally, it is important to notice that phosphonic acid derivatives exhibit two pKa values comprised between that of carboxylic and sulfonic acids. The two acidities are quite different, close to 1.5 and 6–7 for the first and the second one, respectively. These values strongly depend on the structure of the R′ group (Scheme 1) and on the electronic effects of the substituents. The acidity of these compounds can be considered as strong and often leads to the attack and dissolution of metals giving the corresponding salts. These phosphonic acids can be linked to metals according to various patterns through mono-, bi- and tridentate (Scheme 2).14 These multilinks have been largely explored for the synthesis of coupling molecules in the area of hybrid materials, for self assembly15 and for the obtaining of monolayers onto metal surfaces.
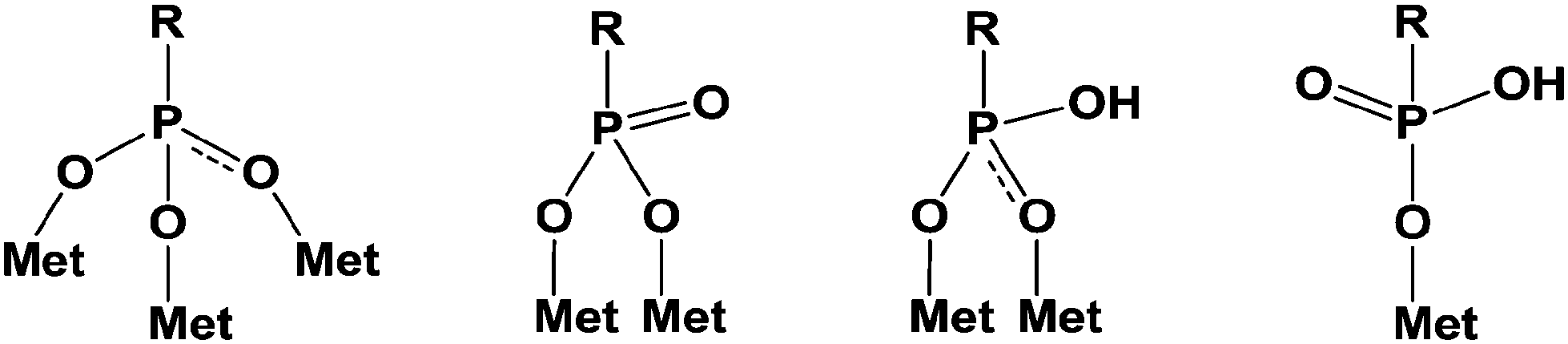 | ||
| Scheme 2 Main linkages for coupling of phosphonates with metal oxide (Met). | ||
This Review presents relevant work achieved to date on the obtaining of various hybrid materials resulting from the combination of inorganic compounds and organic phosphonated molecules. Applications of such materials will also be considered and some relevant references will be given. Linkage to oxides will be deeply investigated using alumina, silica, and titanium, zirconium, zinc, indium tin oxides. Then, hydroxyapatite and calcium carbonate will be considered. Finally, perspectives on such hybrid materials will be given, focusing on the use of clays as valuable inorganic compounds to produce hybrid materials. The synthesis of phosphonated molecules is not discussed here since this is not in the scope of the Review.
2 Alumina
Alumina particles are widely used for several applications, and the grafting of molecules containing phosphonated moieties proved to be of interest to favor the compatibility and the dispersion of particles with organic polymers. Phosphonic acids and their related phosphonates can be linked to alumina particles with strong and covalent Al–O–P bonds. Usually, the grafting of phosphonic species onto inorganic particles is easier than other functions like carboxylic groups. A wide range of examples dealing with the use of phosphonated low molecular weight molecules, oligomers or polymers has been described in the literature and the most famous examples are hereafter described. Several authors investigated the linkage of alkylphosphonic acids and derivatives bearing heteroatoms or other functional groups with alumina. Beside simple surface modifications of mineral particles, aromatic structures, functional polymers or original structures have also been linked onto alumina particles in order to lead to hybrid materials conferring new properties and applications to alumina.2.1 Grafting of alkylphosphonic structures and derivatives
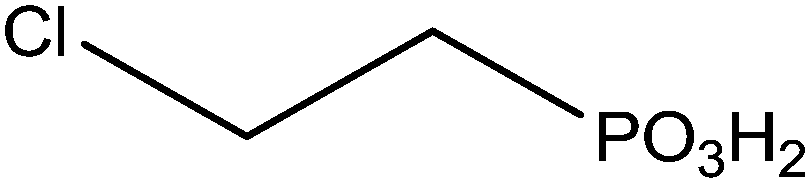
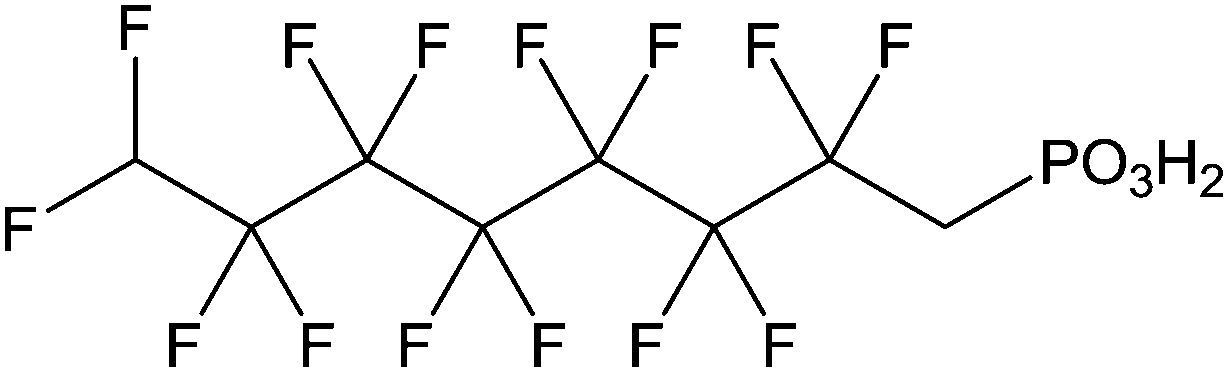
The grafting of alkylphosphonic acids or derivatives has been widely described in the literature.16–20 These molecules are made by a linear alkyl chain bearing a phosphonic acid group as chain end (CH3(CH2)nP(O)(OH)2). After grafting, the obtained hybrid material got new properties such as hydrophobicity or corrosion resistance. Indeed, the grafted layer led to an important increase of the contact angle (≥110°) and induced corrosion resistance of aluminum in acidic media. It was shown that the phosphonic acid headgroups were bound to alumina surfaces via different modes, including mono-, bi- and tridentate. Alkylphosphonic ester derivatives were also used for grafting. For instance, Roskamp et al. described the formation of hybrid materials based on aluminum oxide substrate and phosphonic modified tethered bilayer lipids.21 These novel materials were very interesting for modular construction of model membrane architectures. Some authors also inserted heteroatoms such as fluorine,22 sulfur,23 or chlorine24 into the alkyl chains and compared the properties of these modified structures with non modified alkylphosphonic acids. Brukman et al.22 studied self-assembled monolayers (SAMs) obtained from the grafting of different phosphonated molecules onto alumina particles, notably comparing an alkylphosphonic acid (Scheme 3a) with a fluorinated alkylphosphonic acid (Scheme 3b). Static and advancing contact angle measurements logically revealed an increase of the hydrophobicity of the surface when the fluorinated compounds were used. Indeed, static contact angle measurements were assessed on modified amorphous alumina and gave an increase from 109° to 121° for the fully hydrogenated CH3(CH2)17P(O)(OH)2 and the semifluorinated CF3(CF2)7(CH2)11P(O)(OH)2, respectively. Faure et al.25 described the synthesis of a fluorinated agent (Scheme 3c) able to graft onto hydrophilic alumina to get hydrophobic foaming particles. Indeed, after grafting of the fluorinated phosphonic acid, the resulting material became sufficiently hydrophobic to migrate to the air–water interface and foams could be generated for concentrated dispersions. The increase of the hydrophobicity induced by the presence of the fluorinated agent onto the alumina surface led to an excellent foamability. In another example, Ilia24 synthesized hybrid materials via the grafting of 2-chloroethyl phosphonic acid (Scheme 3d) onto alumina particles. Then, they were incorporated into unsaturated polyester resins, leading to materials with interesting flame retardancy properties. The authors compared the effect of the various particles used and observed better performances with alumina particles compared to silica. The increase in flame retardancy can be explained by synergistic effect between alumina, phosphorus and chlorine. Finally, the last example dealt with the insertion of sulfur atoms23 into the alkylphosphonic acid chains (Scheme 3e). These structures were grafted onto alumina substrates via the phosphonic acid headgroup and X-ray photoelectron spectroscopy (XPS) studies were performed in order to show the influence of sulfur on the chemical stability and homogeneity of the grafted layer. It was demonstrated that the C–S–C bonds were weak, which resulted in a bad film quality evaluated by electrochemical studies. It was assumed that the grafting step probably led to bond cleavages despite mild grafting conditions.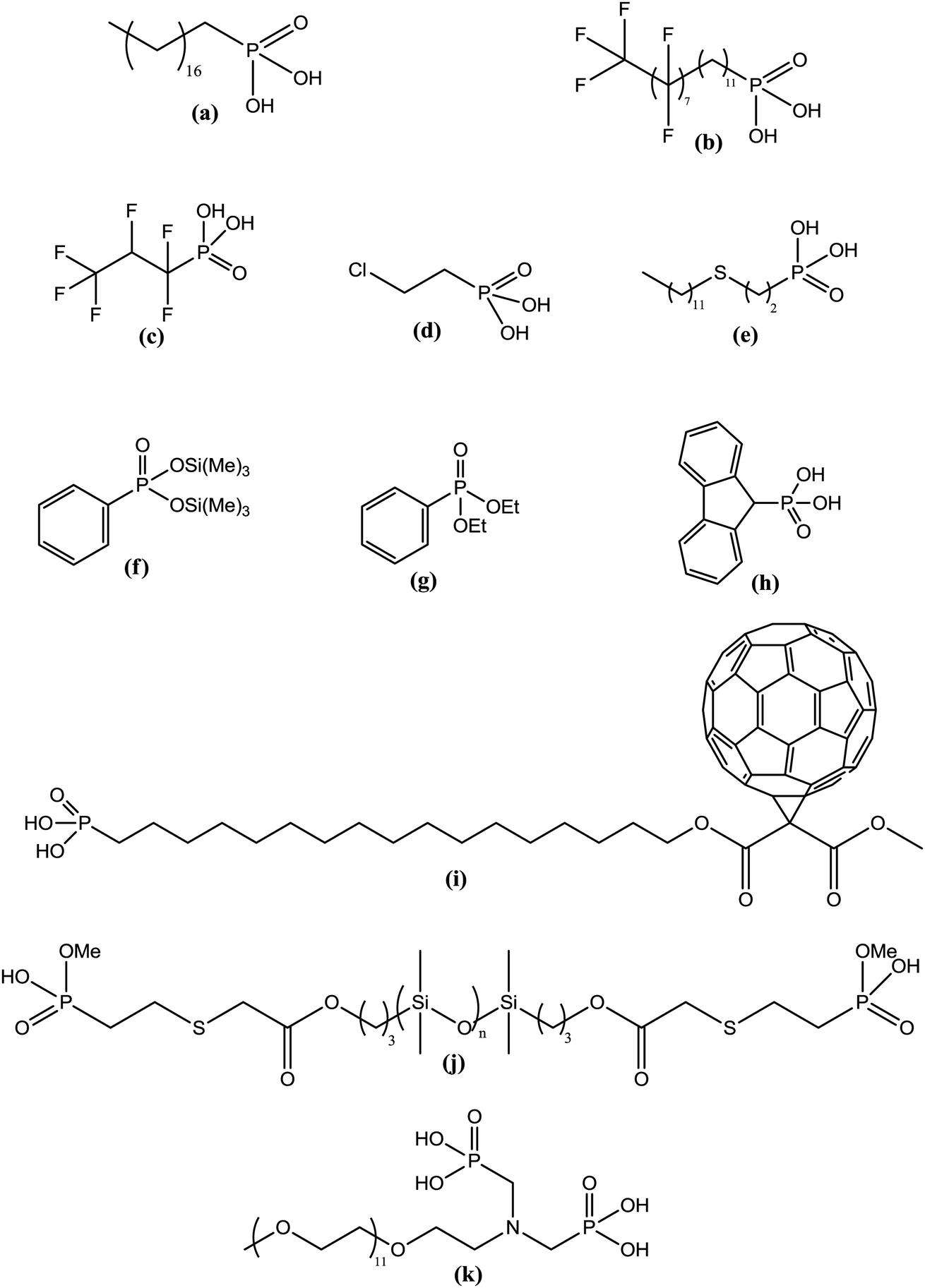 | ||
| Scheme 3 Various phosphonic acid-based structures grafted onto alumina particles. | ||
2.2 Grafting of aromatic-based phosphonic and derivative structures
The grafting of phosphonic derivatives bearing aromatic moieties has also been described in the literature and the anchoring properties of these phosphonic groups studied.26,27 In some examples, the authors compared the use of phenylphosphonic acids with that of phosphonates to modify alumina particles.Guerrero et al.28 studied the modification of alumina through the grafting of, on the one hand, phenylphosphonic acid and, on the other hand, its organic soluble ester derivatives (diethyl phenylphosphonate and bis(trimethylsilyl)phenylphosphonate) (Scheme 3f and g). The success of the organic moieties anchoring was first demonstrated by both 31P and 27Al NMR spectroscopies. The authors showed that the use of phenylphosphonic acid in aqueous solution at pH 6 and at room temperature favored the grafting to the surface of alumina particles, as opposed to the formation of aluminium phosphonate phases. They also noticed the formation of bulk phosphonates phases at lower pH when heating was achieved which could be explained by the solubility of alumina. Such phases were also observed in organic media with phenylphosphonic acid or its bis(trimethylsilyl) ester derivative. The use of diethyl phenylphosphonate allowed controlled grafting of the surface, excluding the formation of bulk phases even under prolonged heating. Thus, organic-soluble dialkylphosphonate derivatives may be useful coupling agent alternatives to low soluble phosphonic acids. A similar work was achieved by Villemin et al.29 who used phosphonate ester headgroups for the grafting under microwave irradiations of different organic moieties including aromatic species (Scheme 3h). In such case, the grafting yield was better for the phosphonates esters in comparison with the corresponding acid derivatives. Additionally, the presence of phenyl groups on the alumina particle surface led to an increase of the hydrophobicity of the hybrid particles. The study of the grafting mechanism has been investigated by some authors30 who concluded that the anchoring of phosphonic acids or phosphonate derivatives was a combination of two mechanisms. The first one deals with the condensation of the P–OH with the Al–OH alumina surface groups. This reaction occurs for several metal oxides because phosphonic acids are stabilized when they are grafted onto metal oxide surfaces. The second mechanism involves the coordination of the phosphoryl oxygen to metallic Lewis acid sites. The grafting of the phosphonic acids or esters can result in the formation of a mono-, bi- or tridentate depending on the structure of the coupling agent and the experimental conditions. Laiti et al.31 described the acid/base complexation properties of phenylphosphonic acid onto boehmite which is an aluminium oxide hydroxide (γ-AlO(OH)) mineral. In this case, they identified mono- and bidentate complexation modes depending on the stoichiometry (phenylphosphonic acid/boehmite). Furthermore, Tsud et al.32 investigated the synthesis of self-assembled monolayer (SAM) based on phenylphosphonic acid and amorphous alumina films obtained by vapor phase deposition in ultra high vacuum. The grafting mechanism was investigated by XPS and indicated the presence of phosphorus tridentate coordination on the substrate surface. Finally, the grafting of alkylphosphonic acid including a fullerene (C60) group (Scheme 3i) was also reported to produce SAMs usable in organic transistors.33 The molecular arrangement of C60 monolayers enhanced conductivity properties of alumina particles leading to better electrical device performances.
2.3 Grafting of phosphonic acid-based polymers
The grafting of polymers onto metal oxide particles proved to be a very interesting way to confer new properties to inorganic phases. Several examples have been detailed in the literature to prepare hybrid materials based on polymer moieties considered as films. Two main strategies have been developed: (i) the “grafting from” route, corresponding to the polymerization of functional monomers directly from the surface of the inorganic particles and (ii) the “grafting onto” route, dealing with the grafting of a functional polymer onto the particle. Both strategies have been widely described in the literature and the main advantages and drawbacks of each method are listed in Table 1. In general, the “grafting onto” method leads to a weak grafting density and layer thickness but the chemical composition of the latter is better controlled. On the reverse, the “grafting from” allows a higher grafting density but the film chemical composition cannot be perfectly controlled.| “Grafting onto” route | “Grafting from” route | |
|---|---|---|
| Scheme |  |
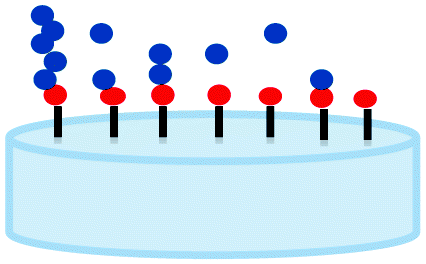 |
| Advantages | Control of the polymer molar mass | High grafting density |
| Control of the chemical composition | High film thickness | |
| Drawbacks | Low grafting density | Low control of the polymer molar mass |
| Low film thickness | Low control of the chain polymer dispersity |
Cinausero et al.34 described in 2008 the modification of alumina particles by different polymers such as polyethylene terephthalate (PET), terethane (TER) or polydimethylsiloxane (PDMS) containing phosphonic acid (Scheme 3j) according to the “grafting onto” process. Nanocomposites based on the obtained hybrid materials were prepared by melt-blending the modified minerals with a poly(methyl methacrylate) (PMMA) matrix. It was shown that the thermal stability and fire behavior of PMMA were improved due to the presence of the nanometric alumina. Significant enhancements of properties in relation with the grafting of the mineral were only noticed for the PDMS phosphonic acid-based formulation. Indeed, the authors assumed that this compound could act in the condensed phase due to its important thermal stability and could also promote modifications of the degradation pathway of PMMA in the interphase region surrounding the alumina particles. Another example dealt with the synthesis of a hybrid material composed of boehmite (AlO(OH)) particle films and of alpha-functional poly(ethylene oxide) (PEO) (Scheme 3k).35 Biofouling applications were tested and the results showed an enhanced resistance to protein adhesion when increasing PEO surface density.
The “grafting from” approach was also investigated. Polymerization of ethylene glycol methacrylate phosphate (EGMP) monomer36 from the surface of alumina nanoparticles was achieved and thermal properties of PMMA and PS nanocomposites containing modified nanometric alumina particles were determined. An important improvement of thermal stability and fire behavior of the obtained nanocomposites was noticed. Another interesting work deriving from the “grafting from” and the “grafting onto” methods was published by Pahnke et al.37 which described the immobilization of benzophenone phosphonic acid on alumina surface (step 1) and the subsequent covalent attachment of different polymers such as polystyrene (PS) and poly(methyl methacrylate) (PMMA) by photoglueing (step 2). The second step was divided in two parts: the first one consisted in the physical deposition of a polymer onto the functionalized surface and the second one in UV-illumination allowing the covalent attachment between UV activated benzophenone and adjacent polymers (Fig. 1). This original way associated the benefits of the two main grafting methods. The formation of such films combining organic and mineral phases could find various applications ranging from protective coatings, to improvement of the biocompatibility of materials, and the fabrication of electronic devices, for instance.
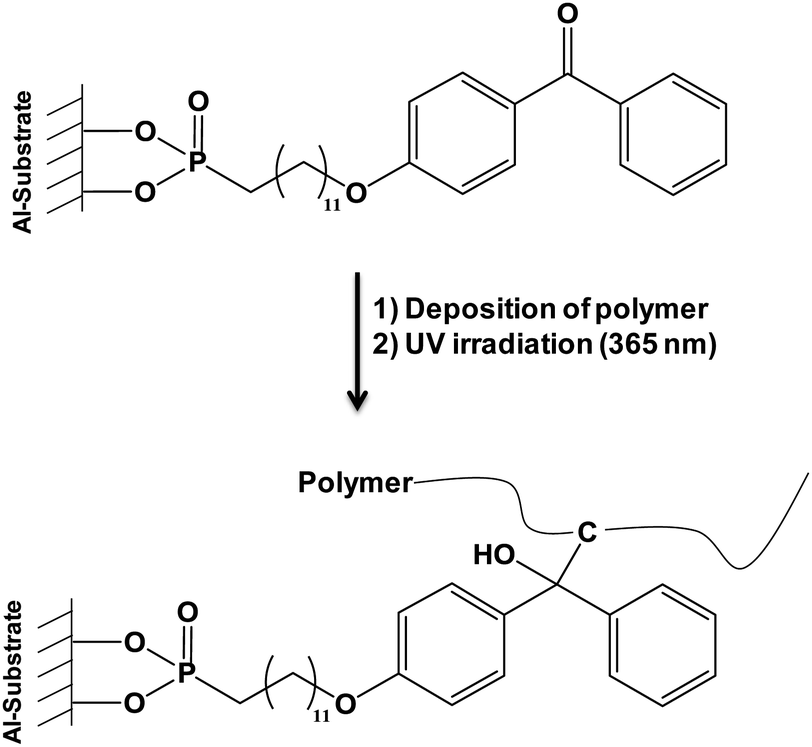 | ||
| Fig. 1 Covalent attachment of polymers onto aluminum-bound benzophenone phosphonic acid.37 | ||
In summary, the reactivity between organophosphonic acids and aluminols is high, leading to the versatile preparation of hybrid materials. The efficient grafting of alkylphosphonic acids, phenylphosphonic acids or functional polymers has been widely investigated towards the formation of hybrid materials allowing different applications including flame retardancy, thermal resistance or organic transistors.
3 Titanium oxide
The first recent work on the grafting of phosphonic acid onto titanium oxide was published by Gao et al. in 1996.38 This paper became a reference, this area of research found more and more interest and the number of publications grew drastically. The two main fields of application of phosphonated titanium oxides are biomaterials and electronics. This part devoted to titanium oxide describes the main studies focused on the grafting of phosphonic acid (simulation and characterization), and biomaterial or electronic applications of modified titanium oxide materials.3.1 Grafting of phosphonic acid onto titanium oxide
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O, P–O–H, P–O–Ti and TiOx bonds by this technique. They both concluded on the predominance of tridentate bonding mode of phosphonic acid onto titanium oxide. Brodard-Severac et al.42 also observed bidentate species and quantified the rate of each of them: Ti–O–P (80%), PO (15%) and P–O–H (5%). These ratio suggested the preponderance of tridentate. The second most used technique to highlight the grafting was infrared spectroscopy (IR). Most authors used this method since it easily permitted to reveal the presence of organic molecules onto the surface. The disappearance of P–O–H and PO bonds was noticed in favor of a large P–O–Ti signal (between 900 and 1200 cm−1).41 So, the principal grafting mode was tridentate but a shoulder at 900 cm−1 indicated the presence of residual P–O–H and as a consequence some bidentate species. The same result was observed on ZrO2 but the presence of a strong stretching band on pristine TiO2 spectrum hid all the signals of the grafted phosphonic acid.38 These two examples showed that valuable information could be extracted from infrared spectroscopic technique. Quantitative measurements are theoretically possible but technically hard to achieve.
O, P–O–H, P–O–Ti and TiOx bonds by this technique. They both concluded on the predominance of tridentate bonding mode of phosphonic acid onto titanium oxide. Brodard-Severac et al.42 also observed bidentate species and quantified the rate of each of them: Ti–O–P (80%), PO (15%) and P–O–H (5%). These ratio suggested the preponderance of tridentate. The second most used technique to highlight the grafting was infrared spectroscopy (IR). Most authors used this method since it easily permitted to reveal the presence of organic molecules onto the surface. The disappearance of P–O–H and PO bonds was noticed in favor of a large P–O–Ti signal (between 900 and 1200 cm−1).41 So, the principal grafting mode was tridentate but a shoulder at 900 cm−1 indicated the presence of residual P–O–H and as a consequence some bidentate species. The same result was observed on ZrO2 but the presence of a strong stretching band on pristine TiO2 spectrum hid all the signals of the grafted phosphonic acid.38 These two examples showed that valuable information could be extracted from infrared spectroscopic technique. Quantitative measurements are theoretically possible but technically hard to achieve.Quantitative measurements of grafting yields were often achieved by monitoring the phosphorus (or carbon) rate thanks to elementary analysis. Variations of P content depending on phosphoric species was found but more than one monolayer grafted on all samples was never observed.41 The carbon percentage was also used to determine the surface coverage.38 Knowing the area of one grafted phosphonic acid (24 Å2) onto the surface, TiO2 was found to be only covered at 61%. With exactly the same method, Pawsey et al.46 determined a grafting close to that of a long alkyl chain phosphonic acid monolayer. On the other hand, with short alkyl chains, the coverage was lower (around 80% of the surface). This technique was also applied in another study which concluded on the possibility to form well ordered monolayers onto titanium oxide with grafting density equivalent to a model structure obtained by grafting with trichlorosilane.47 A part of the literature dealt with self-assembled monolayers and the most used technique to characterize the modified surfaces was atomic force microscopy (AFM). A spray coating technique was used to graft phosphonic acid onto TiO2 support and AFM picture showed islands dispersed onto the surface.45 The thickness of all islands was similar and corresponded to the height of a monolayer. In-depth X-ray photonic spectroscopy (XPS) study of phosphonic acid grafting was also performed.48 The authors grafted a phosphonic acid with a C11 alkyl chain terminated by a thiophene group (Scheme 4a). A lot of information could be extracted from the XPS spectra such as the orientation of the molecules at the surface, the thickness of the layer (2 nm) and the bonding mode of the phosphonic acid. For titanium oxide, the authors found a layer specifically bonded by phosphonic acid through a tridentate mode. Lastly, Ruiterkamp et al.49 functionalized titanium oxide nanoparticles by long aliphatic phosphonic acid. The authors found better particle coverage after two cycles of grafting treatment. The second grafting cycle permitted to obtain transparent dispersion in toluene without any aggregation which was not the case with only one step. When introduced in poly(benzyl acrylate), these modified particles raised the refraction index of the polymer and gave transparent films. The second grafting cycle was performed by a phosphonic acid bearing a vinyl end group (Scheme 4b) which enabled further modifications (such as thiol–ene reaction).
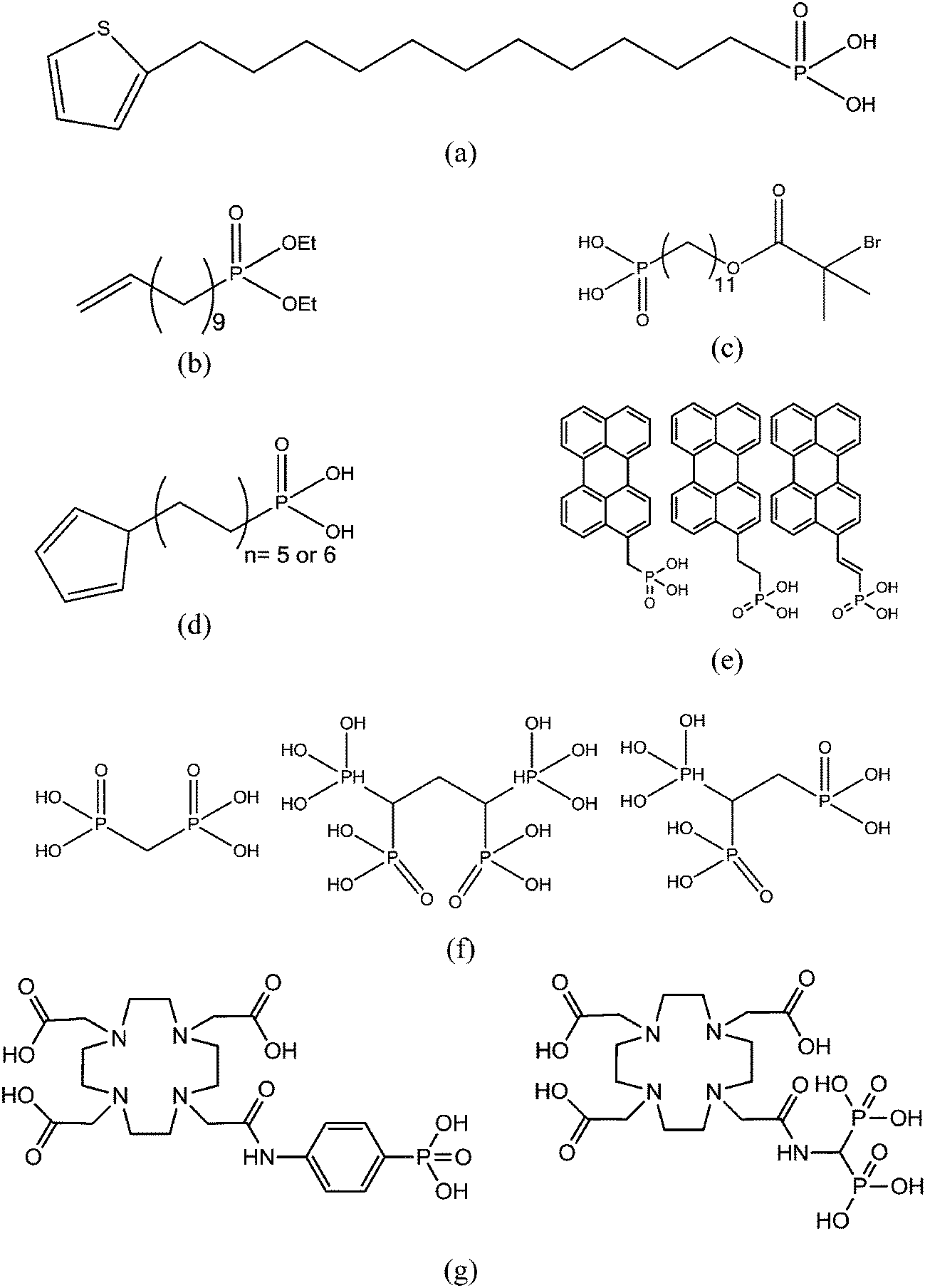 | ||
| Scheme 4 Chemical structures of phosphonic acids grafted onto TiO2. | ||
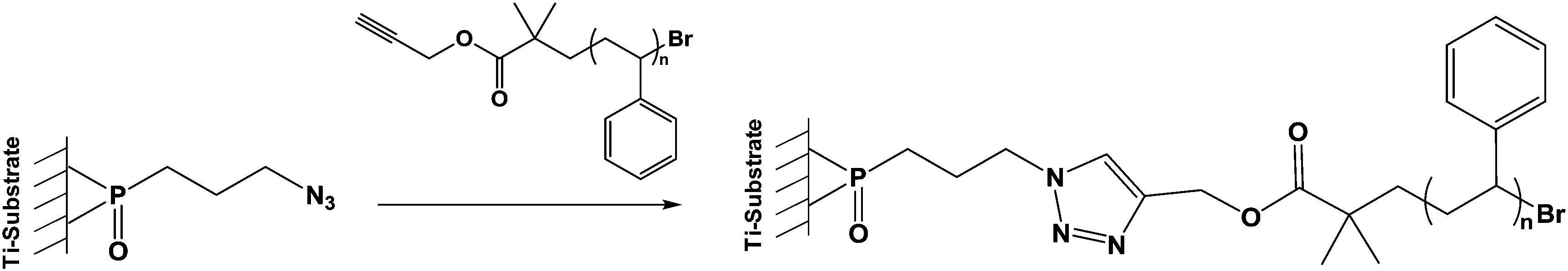 | ||
| Scheme 5 Click reaction of an alkyne terminated polystyrene with an azide phosphonic acid grafted onto TiO2 particles. | ||
Oberoi et al.52 studied adhesion between conducting polymers and inorganic electrodes. First, a phosphonic acid bearing a pyrrol group (Scheme 4d) at the chain end has been grafted onto titanium oxide. In a second step, these grafted pyrrol entities have been polymerized to give polypyrrole. The obtained polymer layer was strongly linked to the oxide and revealed a better oxidation potential. Zoulalian et al.15,53 studied the grafting of a phosphonate-based terpolymer for antifouling applications. This polymer was composed of three different methacrylates: an alkyl phosphonated methacrylate, a butyl methacrylate and a methacrylate bearing a polyethylene glycol chain (Mn = 2000 g mol−1). The synthesis was performed by a conventional free-radical polymerization process. The organization of the grafted terpolymer onto the TiO2 surface was ascertained by XPS measurements. The grafting was provided by the phosphonic moieties and the polyethylene glycol chains formed brushes at the top of the surfaces. The grafted terpolymers show better stability facing pH from 2 to 9 than usual existing systems. Excellent resistance to protein adsorption was achieved when exposed to full human serum.
3.2 Applications
In summary, phosphonic acids can be strongly bonded to titanium oxide by Ti–O–P bonds and the resulting grafts showed a good resistance to hydrolysis. Titanium oxide-based hybrid materials proved to be useful for different kind of applications, including electronic devices and biomaterials. In the latter case, an important advantage using multi phosphonic acid group molecules is the opportunity for phosphonic acid not involved in the grafting to take part to osteogenesis.
4 Zirconium oxide
Zirconia or zirconium oxide have found many applications in industry. The main property of this oxide is its thermal resistance and hardness making it notably interesting for engine parts, or insulators. Zirconia also possesses a larger band gap than silicon oxide interesting for dielectric devices. Actual researches on the grafting of phosphonic acid groups onto zirconia mainly deal with the characterisation of the grafting efficiency and properties of the modified particles (physical, mechanical and dielectric properties, etc.).4.1 Study of the grafting alkylphosphonic acid molecules
Gao et al.38 described the grafting of octadecylphosphonic acid onto various substrates: TiO2, Al2O3 and ZrO2. Two different zirconium oxide substrates were evaluated, zirconia powder (40 m2 g−1) and silica coated by zirconium oxide with a greater specific area (100 m2 g−1). Probably due to a lack of stability, zirconated silica showed weak surface coverage (26%) and formation of zirconium-phosphate compound. On the contrary, phosphonic acid was strongly bonded to pristine zirconia by tridentate bonding mode (highlighted by phosphorus NMR spectroscopy) and led to a good surface coverage (73%). The authors claimed the formation of a monolayer of phosphonic acids similarly to the usual case of self assembled monolayer onto planar metallic surfaces. On a theoretical point of view, it was shown that the ordered organisation of the monolayer was closer from a bilayer model system rather than a crystalline polymer model. In a second paper,61 the same authors highlighted the role of the surface roughness in the creation of defects in the self-organized monolayer. These defects improved the motion of alkyl chains and were also responsible of the decrease of the order-disorder transition temperature. Two papers were published in 2011 on making more densely packed self-assembled monolayer of phosphonic acid (using acid phosphonic acid, phosphoric acid, and bis-phosphonic acid) onto zirconium oxide. In the first paper, it was shown that varying the alkyl chain length had an impact on the surface coverage because shorter chains did not tend to self-assemble, leading to unreacted hydroxyl groups at the surface.62 For higher chain length, van der Waals interactions between the alkyl spacers increased, allowing the molecules to self-assemble, yielding densely grafted layers. It was also demonstrated that the variation of the anchoring group had an impact on the alkyl chain ordering. In the second paper, the influence of the curvature of the substrate on the alkyl chain ordering was highlighted.63 The more plan was the substrate, the more organized was the monolayer. Phosphoric acid, phenylphosphonic acid, and octyl phosphate were also covalently grafted to modify the polarizability of zirconia particles.64 Zirconia with high degree of crystallinity was first prepared to improve its stability in acidic conditions. This resistance was proved because no changes in the specific area and crystallinity were observed after the grafting treatment. Moreover, no zirconium phosphate phase was found in the material proving the resistance of the oxide to phosphonic acid. Additionally, the 31P NMR spectrum showed three general bonding modes: two different bidentate modes and one tridentate mode. Another study reported the hydrogen bonding of grafted carboxyalkylphosphonate groups by solid-state 1H NMR. The latter also demonstrated the tridentate bonding mode of phosphonic acid with zirconium oxide because only few P–OH groups remained.65 Finally, Marcinko and Fadeev66 studied the resistance to hydrolysis of different grafted anchoring agents onto zirconia. At ambient temperature, grafted anchors remained unchanged. At 65 °C compounds grafted from monochlorosilane was completely desorbed in a few hours. The grafted tridentate silane (–SiH3) had better stability at 65 °C probably due to a self-polymerization at the surface. The best result was obtained with grafted phosphonic acid. Indeed, only 2–5% of the grafted acid was cleaved out of the particle surface after one week at pH 1–10.4.2 Applications of grafted zirconia
Only few publications described the used of grafted zirconia to improve properties of the pristine oxide. Limited interactions between proteins and zirconium oxide proved to be of interest in biochemistry to improve separation steps like in chromatography or ultrafiltration. Randon et al.67 modified titanium oxide and zirconia-based ultra-filtration membranes with short alkyl phosphonic acid (phosphoric acid, methyl, ethyl, butyl, and phenyl phosphonic acid). Modified oxides exhibited the same electrostatic charges but possessed better rejection rate and flux than unmodified membranes. Ethylenediamine-N,N-tetramethyl phosphonic acid (EDTPA) (Scheme 6), a molecule similar to EDTA, was also used to modify zirconia surface.68 EDTPA modified zirconium oxide was tested as stationary phase for chromatography with better selectivity and mass recovery of proteins. Authors explained this improvement by the decrease of the interactions between proteins and zirconia due to the EDTPA grafting. Unfortunately, the grafting was not stable at low pH. As a result, columns only worked for neutral and basic proteins.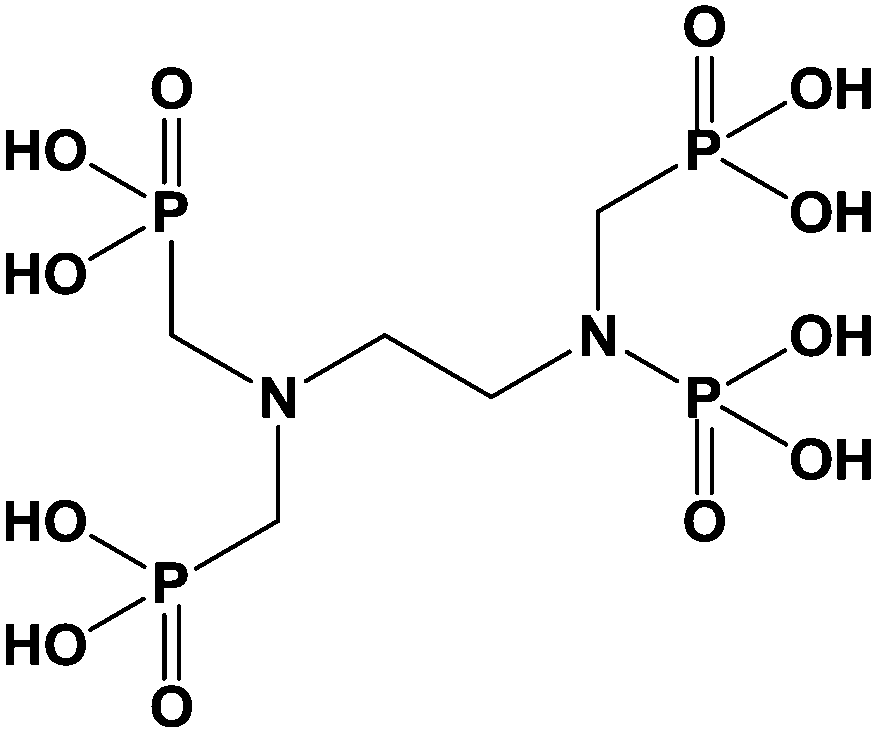 | ||
| Scheme 6 Chemical structure of ethylenediamine-N,N′-tetramethylphosphonic acid (EDTPA). | ||
Zirconia exhibited good mechanical properties (hardness) making them interesting as reinforcing filler. Like silica, they had to be modified to achieve a good dispersion in usual polymers. Nevertheless, zirconium oxide is an expensive mineral, so not really competitive for industrial applications. Sajjad et al.69 incorporated functionalized nanoparticles of zirconia in epoxy resin. The dispersion of particles was improved by the grafting of polyethylene oxide thanks to phosphonic acid anchoring group. Rheological measurements showed a great increase of the rubbery region modulus by the incorporation of the modified particles. On the contrary, no changes were observed on the thermal stability of the resulting material. Finally, Ha et al.70 synthesized an original hybrid material for dielectric film applications. They first deposited zirconia onto a silicon wafer and then self-assembled a monolayer of an aromatic conjugated compound bearing a phosphonic acid at one chain end and two hydroxyl groups at the other end. The authors then generated a second layer of zirconia on the modified surface thanks to these hydroxyl groups. The process described here was then repeated from one to four times to obtain multilayers. The resulting hybrid material showed high electrical capacitance, low leakage current density and excellent thermal stability.
To sum up, zirconia grafted with phosphonic acid-based compounds has not been widely studied but the few works devoted to this grafting showed a great potentiality for this surface modification. Indeed, the quoted publications essentially reported a tridentate grafting which is the strongest way to link phosphonic acid onto a mineral substrate. Additionally, the paper of Marcinko and Fadeev66 pointed out the remarkable strength of this link leading to high resistance to hydrolysis of the modified zirconia.
5 Zinc oxide
Zinc oxide (ZnO) is a transparent piezoelectric metal oxide with potential applications in sensors,71 light-emitting diodes72 (LEDs), lasers,73 transistors, and solar cells.74 ZnO is a direct wide-gap semiconductor with specific properties including high electron mobility and strong luminescence. The ZnO surfaces are often used for optoelectronic applications, and various nanoobjects have been successfully synthesized such as nanowires,75 nanorods,76 nanobelts,77 or nanotetrapods.78 The functionalization of zinc oxide can be achieved by using different anchoring organic molecules, oligomers or polymers bearing carboxylic acid,79 phosphonic acid,80 organosilane81 or thiol81 groups. Several authors described the grafting conditions of phosphonic acid anchors onto ZnO and the type of linkage formed between the hydroxyl groups of the metal oxide and the acid functions. Smecca et al.82 reported a theoretical and an experimental study on the grafting of propylphosphonic, monofunctional tetradecylphosphonic, bifunctional 3-phosphonopropanoic, and 16-phosphonohexadecanoic acids. Spectroscopic (FTIR and XPS) characterization and DFT modeling permitted to understand the interactions which drove the surface anchoring of phosphonic acids on ZnO surface. This study highlighted a multidentate coordination involving PO and P–OH bonds and confirmed the higher stability of phosphonic acids grafts in comparison to carboxylic acids ones (Fig. 2). The multidentate anchoring was confirmed by first-principles density-functional total energy calculations.83 Hotchkiss et al.84 reported a comparable study concerning the linkage of phosphonic acids onto ZnO surface and the packing of the alkylphosphonic chains (octylphosphonic, octadecylphosphonic, and (3,3,4,4,5,5,6,6,7,7,8,8,8-tridecafluorooctyl)phosphonic acids).
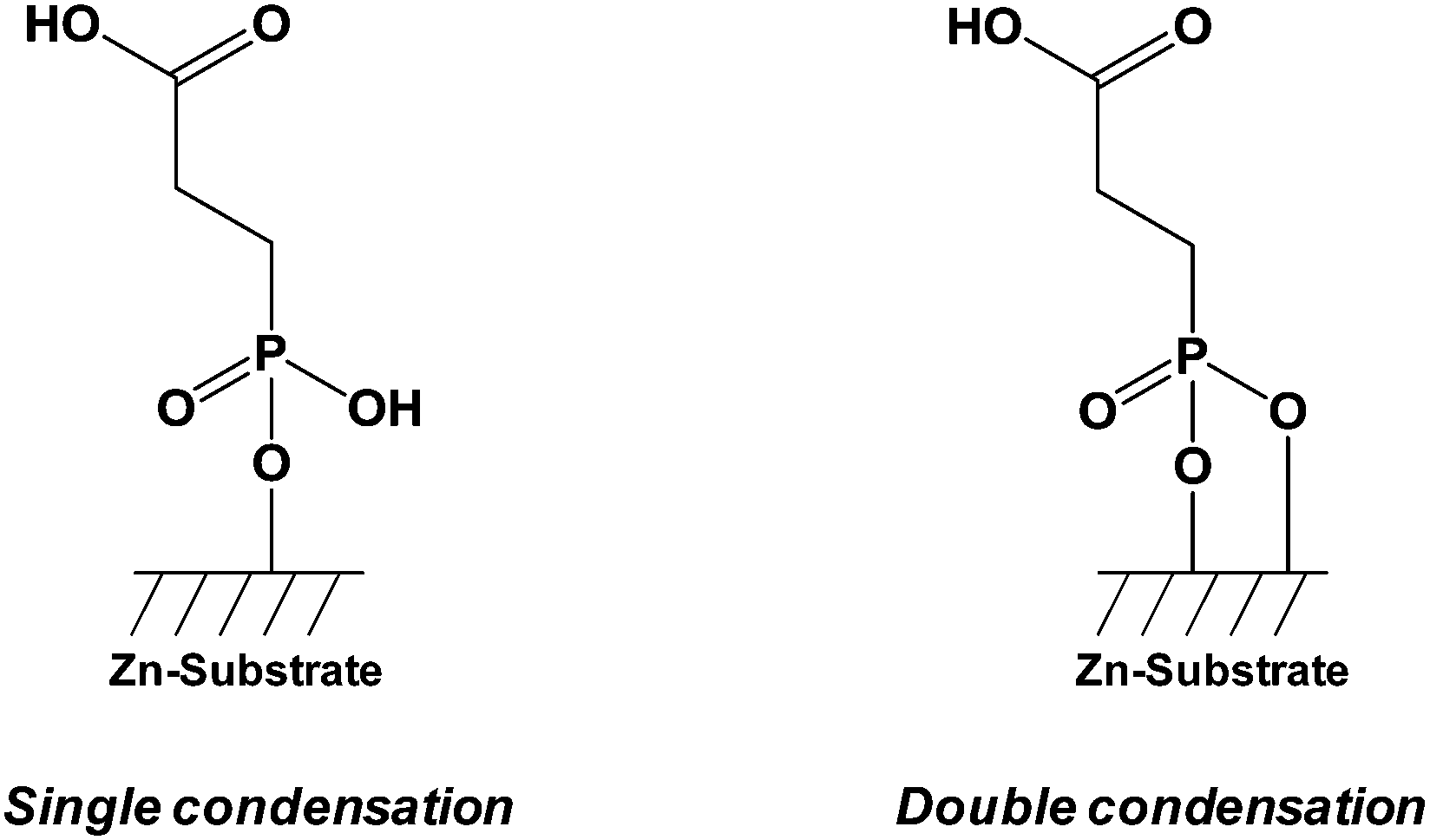 | ||
| Fig. 2 Representation of the interactions between 3-phosphonopropanoic acids (CPPA) and ZnO substrates.82 | ||
Characterizations were achieved by X-ray photoelectron spectroscopy (XPS), infrared reflection-absorption spectroscopy (IRRAS), atomic force microscopy (AFM) and contact angle measurements. Homogeneous and well-packed monolayers were obtained, with a good surface coverage, and a tridentate coordination.
Zinc oxide is often used for anti corrosion applications and several authors pointed out the interest of phosphonic acids grafted onto metal oxide surfaces for the protection against corrosion attacks. The attachment of organic molecules through thiol functions and phosphonic derivatives was studied with a possible application for protection of ZnO surfaces against chemical attack.85 Indeed, grafting of 1-hexanethiol and 1-hexanephosphonic acid onto ZnO was achieved and the corrosion resistance was tested in the presence of ammonium chloride. The results indicated that a phosphonic acid-anchored alkyl chain provided great corrosion resistance in comparison with thiol-anchored alkyl molecules. ZnO layers are also usually used in solar cell for their good electronic properties and the grafting of specific molecules can improve them. Stubhan et al.86 investigated the functionalization of Al-doped ZnO surface with phosphonic acid anchored self-assembled monolayers (SAM). Different SAMs were tested including aliphatic SAMs and fullerene functionalized SAMs in order to increase solar cells properties. The use of the fullerene SAMs allowed an improvement of the charge transfer and a decrease of the series resistance while the shunt resistance remained high.
In summary, the sorption of organophosphonic acids onto ZnO substrates allowed a good stability of the organic-ZnO interface. The main potential applications for ZnO modified surfaces dealt with the improvement of the protection against corrosion of ZnO surfaces and the enhancement of solar cell properties.
6 Silica
Silicon is the most important semiconductor material in the microelectronic industry especially because of the chemical and electrical stability of the interface with its oxide SiO2.87 Therefore, the potential chemical modification of SiO2 allowed the improvement of these different characteristics and it became a challenge for chemist to modify the surface of this material. Indeed, a lot of works have been devoted to the functionalization of SiO2 by grafting moieties using OH groups present onto the silica surface.88 The grafting of molecules onto silica support needs to take into account different fundamental issues. First, the reaction between phosphonic acids and hydroxyl groups of the silica surface requires high activation energy. Second, the weak chemical stability of the Si–O–Si bond at the interface between the organic phase and SiO2 surface has to be kept in mind.89,90 To date, the most described method allowing an effective grafting onto silica surface is the silanization.91–93 This method was a more effective way than the grafting of phosphonic acid onto the silica. As a result, the phosphonic acid function was not involved in the grafting and was available for the complexation of uranium, for instance. Nevertheless, some examples described the grafting of the phosphonic acid moieties onto silica-based materials. Mutin et al.30 described the selective functionalization of TiO2–SiO2 surface using phosphonic acid derivatives. Grafting of phosphonic acids logically occurred onto TiO2 domain whereas organosilanes, also used in this study, were fixed onto SiO2 layer (Fig. 3). Indeed, as described in the part allocated to TiO2, the phosphonic acid gets more affinity for titanium oxide surface and the Ti–O–P bond is more stable than the Si–O–P one.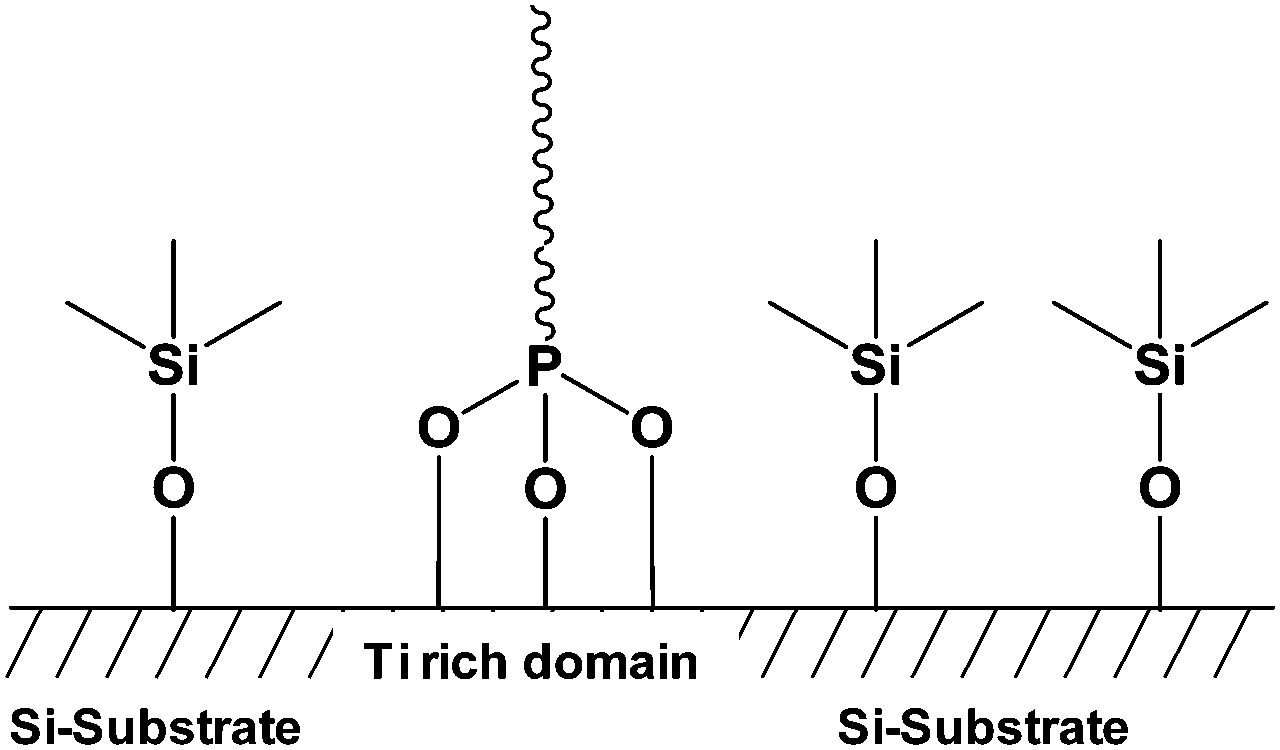 | ||
| Fig. 3 Representation of selective bifunctionalization of the SiO2–TiO2 mixed oxide by phosphonic acid and organosilane.30 | ||
Other works reported the grafting of phosphonic acid-based molecules after the functionalization of SiO2 surfaces with a controlled coverage of aluminum oxide.94,95 In another contribution, an efficient process was developed to spin-cast phosphonate SAMs (from 8-(11-phenoxy)undecoxyoctyl phosphonic acid, n-octadecyl phosphonic acid, and 1H,1H,2H,2H-perfluorododecyl-1-phosphonic acid) onto a SiO2 substrate that was first activated by an in situ generated nanoscale aluminum oxide layer to increase the surface reactivity for phosphonic acid molecule binding.96
Self-assembled organic monolayers were also directly bound to the native oxide surface of Si. Smooth and homogeneous self-assembled monolayers of 2-(thienyl)hexylphosphonic acid on the native oxide surface of silicon were prepared by a process called air–liquid interface-assisted method.97 The prepared SAMs were packed in an orderly manner, and adhered firmly to the substrate surface. Additionally, it is interesting to notice that this simple and fast method only required a small amount of solution. Finally, monolayer films of both aliphatic and aromatic phosphonates (octadecylphosphonic acid, 11-hydroxyundecyl phosphonic acid and α-quarterthiophene-2-phosphonates, respectively) were bound to SiO2/Si by a method called tethering by aggregation and growth (T-BAG).89,98,99 In that case, no pretreatment of the native oxide was required. The substrate was held vertically in a solution of the corresponding organophosphonic acid below its critical micelle concentration (CMC), and the solvent was allowed to evaporate slowly. So, the organophosphonic acid was transferred to the surface. Finally, a heating step was required to lead to the formation of a chemical Si–O–P bond, leading to dense organophosphonate films bonded to the SiO2/Si surface. Growth was not limited by surface OH content, and densely packed surface bound monolayer films were obtained.
In summary, the use of phosphonic acids as anchors for SiO2 substrates is not the optimal choice in comparison with silane groups. The only possibility for grafting phosphonic acids onto SiO2 surfaces is to use the T-BAG method. Nevertheless, some contributions reported the modification of SiO2 with aluminum oxide to activate the surface.
7 Indium tin oxide
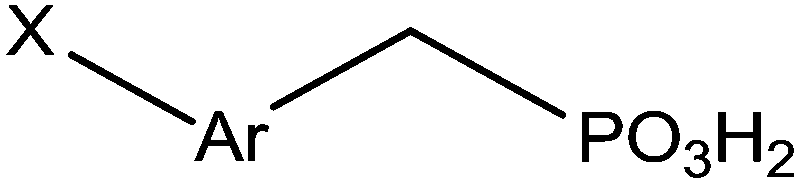
Indium tin oxide (ITO) is one of the most interesting materials for transparent conductors for optoelectronic applications, due to a relatively high conductivity (sheet resistance of 10–20 Ω−1), and transmission (>80%) in the visible region of the solar spectrum.100 Additionally, ITO has a stable chemical structure and can be easily patterned using lithographic techniques which are currently used for device fabrications. ITO films are widely used for transparent conducting materials in liquid crystal displays and get optical and electrical properties leading to various applications.101 In the industry, they are prepared by thermal evaporation deposition or by magnetron sputtering.102 The intrinsic properties of ITO layers are very interesting but they can be improved by the addition of various functionalities. Several anchoring molecules, including carboxylic acids,103 phosphonic acids104 and silanes105 can be used for the grafting of specific moieties. Several studies explained the choice of phosphonic acids for the ITO surface modification.106 Indeed, phosphonic acids can be strongly linked onto ITO supports and are less prone to self-condensation than organosilanes. Moreover, phosphonic acid-based films are more resistant to hydrolysis than those formed from organosilanes or carboxylic acids.38 The most described applications involving the grafting of phosphonic acids onto ITO surfaces concern optoelectronic devices.107 Table 2 reports the different phosphonic acid-based structures grafted on ITO layer, the deposit conditions and the potential applications.| Phosphonic acid-based structure | Deposition method | Applications/properties | Ref. |
|---|---|---|---|
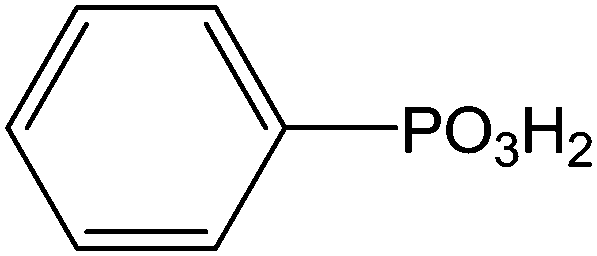 (and derivatives) (and derivatives) |
Immersion in solution during 24 h + heating step | PLED | 110 |
 |
Immersion in organic solution during 16 h + heating step | OLED | 111 |
 |
Immersion in organic solution overnight + heating step | PLED | 112 |
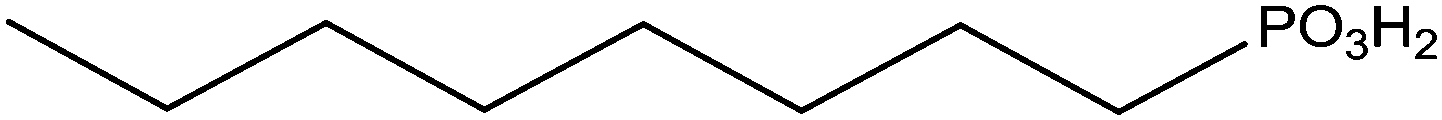 (or fluorinated derivatives) (or fluorinated derivatives) |
Immersion in organic solution during 80 min + heating step | OLED | 113 |
 |
Immersion in organic solution during 12 h + heating step | PLED | 114 |
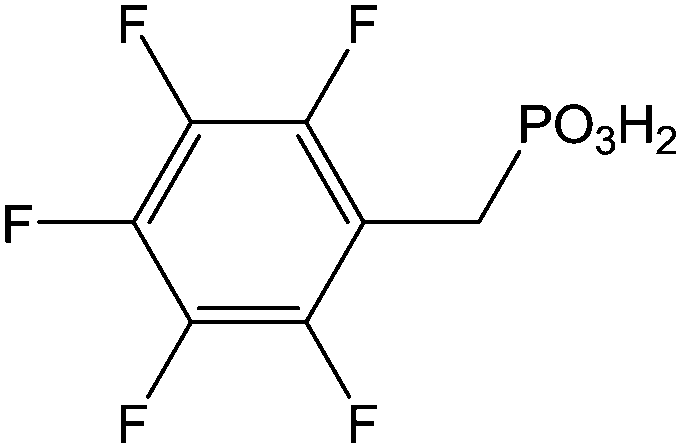 (and derivatives) (and derivatives) |
Immersion in organic solution + heating step | OLED-OPV | 106 |
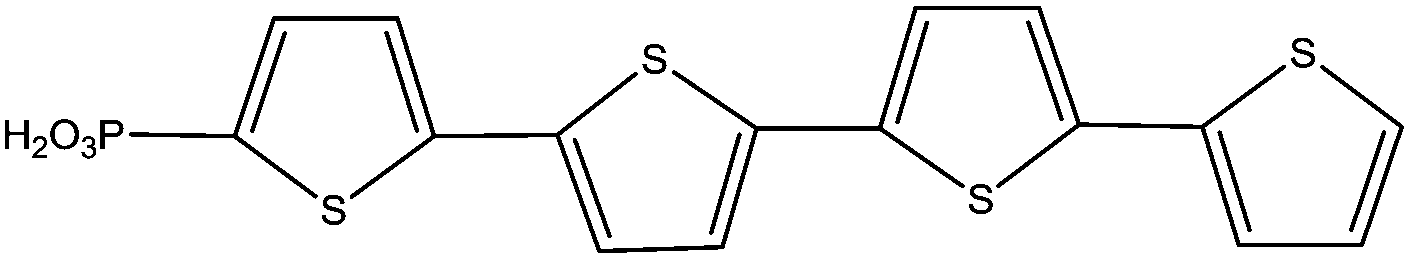 |
Immersion in organic solution + evaporation step | PLED | 115 |
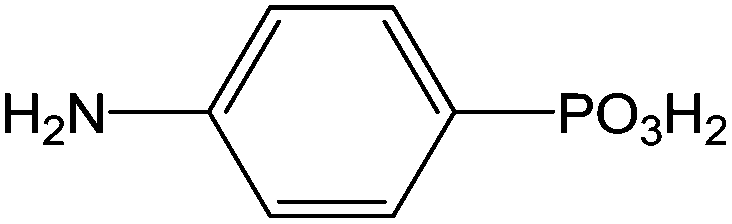 |
Immersion in aqueous solution during 24 h + annealing 120 °C | Biochemical | 116 |
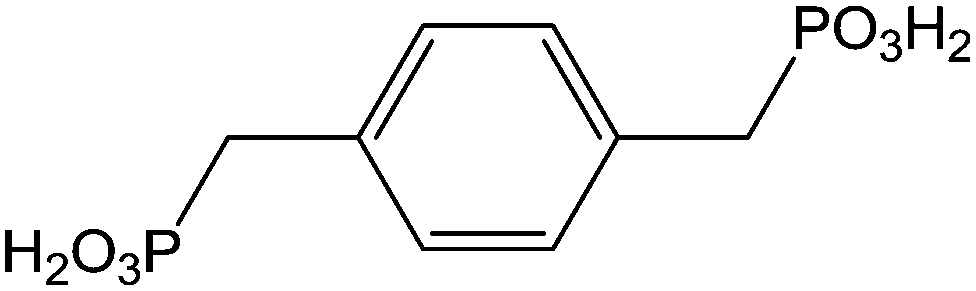 |
Immersion in organic solution + heating step during 48 h | OLED-OPV | 117 |
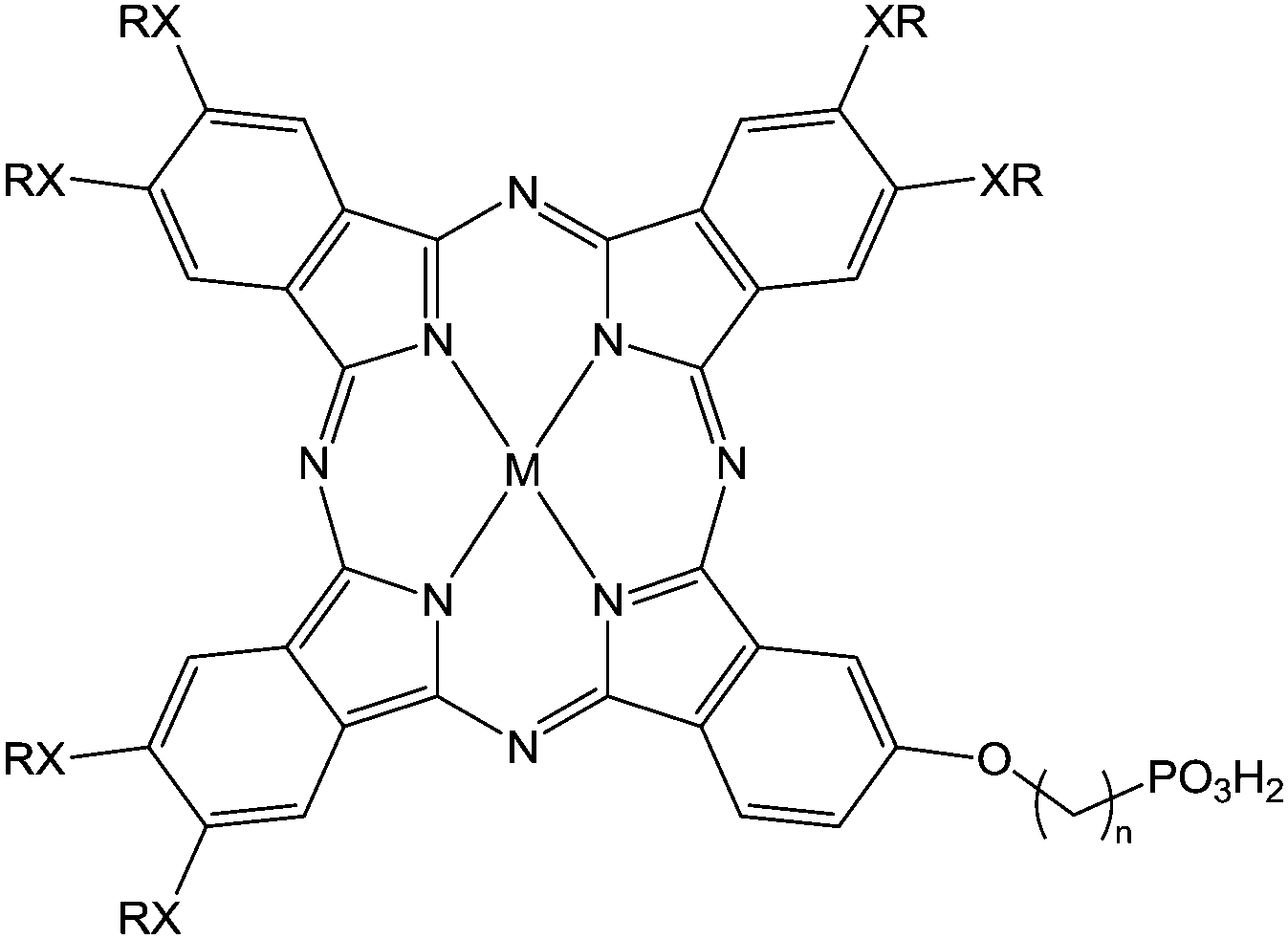 (n = 3; X = O, S; M = H2, Zn) (n = 3; X = O, S; M = H2, Zn) |
Immersion in organic solution during 3 h | OFET-OPV | 118 |
Fundamental studies were led for a better understanding and knowledge of the grafting process of phosphonic acid derivates onto Indium Tin Oxide surface. The impact of the synthesis conditions on the grafting rate was investigated by Chen et al.101 who showed the effect of various solvents on self-assembled monolayers formed from phosphonic acid-based moieties on ITO. Typically, it was shown that solvents of choice should have as low dielectric constant as possible but did not have to complex with the underlying ITO substrate. Other studies dealt with the binding energy and mechanism of organophosphonic acids on ITO. Studies highlighted the bidentate and tridentate binding modes of phosphonic acid106 and proved that the binding energy was stronger for the phosphonate linkage compared to the carboxylic acid one.108 Self-assembled monolayers (SAMs) of alkanephosphonic acids with chain lengths varying from 8 to 18 carbon units were formed on thin films of ITO.109 The direct effect of the phosphonic acid anchoring on the surface hydrophobicity and roughness was investigated. Long-chain SAMs were more highly ordered, and had a smaller tilt angle than short-chain SAMs. It was also shown that when the ITO surface roughness became greater than the SAM chain length, the SAMs became relatively disordered.
Other contributions reported the development of new devices with enhanced properties. As shown in Table 2, optoelectronic applications are mainly targeted as the grafting of specific molecules anchored by phosphonic derivatives allows the enhancement of several properties, mainly optoelectronic ones (work function, current density, brightness or charge injection in the case of light emitting diode devices). A wide range of phosphonic acid-based structures was investigated and allowed the improvement of the characteristics of the semiconductor devices. Indeed, the interfaces between electrodes and organic/polymer materials greatly affected the device performances. Different organic electronic devices are nowadays developed including organic light emitting diodes119 (OLEDs), polymer light-emitting diodes120 (PLEDs), organic field effect transistors112 (OFETs), and organic photovoltaic's121 (OPVs) (Table 2). In such context, the modification of an ITO-based electrode was achieved using alkylphosphonic acids in order to improve OLEDs characteristics.113 Partially fluorinated phosphonic acid was used to treat ITO surfaces and the resulting electrode performances were compared with that of an electrode treated by air plasma. The obtained results indicated a comparable change of work function (5.3 and 5.4 eV with phosphonic acid-based ITO and air plasma treatment, respectively) and a higher stability over time. The phosphonic acids modified ITO allowed an improvement of the lifetime of the resultant OLEDs devices. Indeed, the devices treated with air plasma led to a half-lifetime of 97 hours whereas a phosphonic acid modified ITO device exhibited a half-lifetime of 616 hours. Indium tin oxide supports were also modified with asymmetric phtalocyanines.118 The electrochemical properties of phosphonic acids modified ITO substrates were measured using cyclic voltammetry. These functionalizations involved a significant modification of the thermodynamics and kinetics of the charge transfer. Finally, even if the main applications are related to optoelectronic devices, some authors tried to use modified ITO for other fields described hereafter. Depending on the nature of the grafted molecules, different properties can be reached for ITO substrate. Chen et al.116 grafted 4-aminophenylmethylphosphonic acid onto ITO surface and developed two different synthesis methods, the first one consisting in the self-assembly of these molecules through the phosphonic acid groups and the second one corresponding to the electrochemical reductive adsorption of the aryl diazonium salt. According to these grafting methods, the authors designed electrochemically stable ITO-organophosphonic acid layer-gold nanoparticles (AuNPs) and suggested to use this novel strategy for the generalization of the attachment of metal particles on ITO supports and their use in specific biochemical applications.
In summary, the grafting of phosphonic acid derivates onto ITO substrates has been widely used thanks to the strong interactions between organophosphonic acids and ITO. The sorption mechanism has only been slightly studied and most hybrid materials were tested for the improvement of the optoelectronic properties of light-emitting diodes.
8 Miscellaneous
The grafting of phosphonated molecules was also carried out onto hydroxapatite and calcium carbonate which has been considered in the literature and proved to be of interest, notably for biomedical applications.8.1 Hydroxyapatite
Functional phosphonated molecules have been largely used in the area of biomaterials since then can interact with natural inorganic materials such as bone, or tooth, giving organic/inorganic hybrid systems. In this area, phosphonic acids are prone to favor biocompatibility of synthetic polymers such as poly(meth)acrylic resins with natural hard tissues, opening applications for orthopaedic applications: repairing, fixation or implantation. Phosphonic acids are able to form phosphonate complexes and calcium phosphonate promotes the biocompatibility. In some cases, the growth of hydroxyapatite onto the synthetic surfaces is targeted using some simulated body fluids acting as sources of calcium and phosphate in order to develop compositions close to that of hydroxyapatite.122Many developments have been performed in the area of dental restoration. In this case, phosphonic acids were linked to monomers and mixed with bis-phenol A-based (meth)acrylic resins before being polymerized under UV irradiation. Generally, mono or bi phosphonic compounds are classified in vinylics, (meth)acrylates and (meth)acrylamides, the last being more stable under hydrolytic conditions. It should be too long to describe all the chemical structures that have been designed these last years and only some of them can be cited hereafter (Scheme 7).123–131
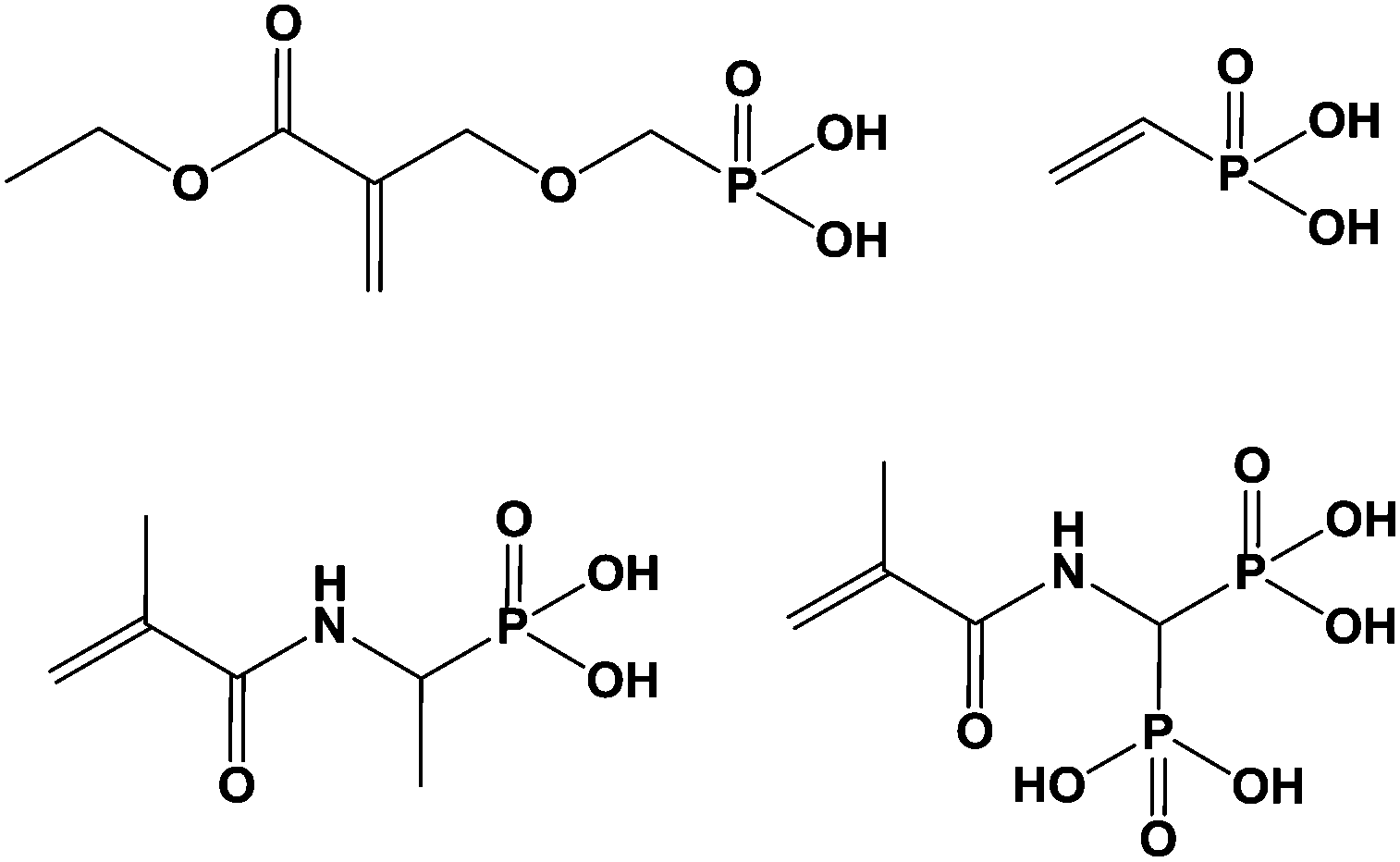 | ||
| Scheme 7 Usual monomers for coupling with hydroxyapatite. | ||
These acidic monomers led to low pH values of aqueous solutions (close to 1.5) and proved to strongly decalcify hydroxyapatite (HAP). HAP treated with these monomers led to calcium salts with modified X-ray diffraction patterns.124 These monomers gave strong adhesion to enamel and dentin and some competitive mechanism between decalcification-adhesion was proposed by Yoshida and co-workers.132 Despite the hydrophilic character of phosphonic acids, the phosphonated monomers were often copolymerized with hydrophilic monomers such as hydroxylethyl methacrylate (HEMA) to obtain hydrosoluble polymers. Bayle et al. investigated the formation of hybrid layers during the use of self-etch adhesives, that is to say, aqueous mixtures of monomers and oligomers able to attack the dentin and to adhere to this natural material through the formation of a hybrid layer.133,134 In this complex layer, after dissociation of HAP and formation of brushite, calcium ions crosslinked polymeric chains thanks to one acidity of phosphonic acids as in ionomer resins. At last, this layer integrated collagen, and mineral crystals linked to polymer chains crosslinked by calcium ions.
8.2 Calcium carbonate
Calcium carbonate (CaCO3) or calcite which is the most abundant mineral is commonly used in various industrial processes. The dispersion of this pigment in plastics or in solution requires to chemically treat its surface. Most often, the cheap stearic acid reagent was used and gave satisfying results. Using phosphonates opened the way to new functionalizing reagents. Unfortunately, the low pKa of phosphonic acids led to a dual mechanism where dissolution/precipitation of CaCO3 took place beside surface functionalization. In some cases, only dissolution/precipitation was observed.135 Some authors also tested the use of phosphonated double hydrophilic copolymers to impact the crystallization of CaCO3 and its final morphology.136The surface modification of calcium carbonate in mild conditions has been slightly studied in the past.137 More recently, Mutin et al. highlighted the formation of dianionic species RPO32− and showed that dissolution/precipitation could not be avoided for long reaction time and high concentration in phosphonic acids.138 Nevertheless, the authors succeeded in functionalizing and analyzing the CaCO3 surfaces.
9 Conclusions and perspectives: use of clays to elaborate hybrid materials
Phosphonate derivatives are efficient compounds for the functionalization of mineral and metal oxides. The ease of synthesizing this class of compounds opens the way to versatile functionalizations starting from simple molecules to (co)polymers permitting the development of smart materials. Furthermore, the stability of phosphonate groups to hydrolysis was proven in the past and makes these groups stable in humid conditions like in biologic fluids. Moreover, phosphonic acids or their ester derivatives can react in aqueous solutions. Due to the low pKa of these acids, the reaction at the surface of the inorganic compounds can be performed in mild conditions, most often at room temperature. It is obvious that this reactivity permits to envisage to link chemically sensitive organic molecules to inorganic surfaces. Besides, phosphorus-based materials are more and more used in the field of flame retardancy. Among possible applications, electronics area remains a field where combining metal oxide with organic molecules should find various developments as proven by the above cited literature.An interesting perspective that has to be considered is the use of phosphonates derivatives for the grafting of bio-based supports. Among all possibilities, clays appear as compounds of choice to produce hybrid materials. To date, the chemical modification of clays was usually achieved by ion exchange reactions or silane grafting. These reactions allowed improving the compatibility and the mechanical properties of composites. In 1996, a first example reported the successful use of mica to produce hybrid materials by grafting phosphonic acid compounds onto this mineral. So, the use of clay appeared as a promising way to produce new materials. Until now, most of the publications focused on self-assembled monolayers (SAM) of fatty phosphonic acids onto cleaved mica and, in few cases, on montmorillonite, kaolinite and halloysite.
Woodward et al. reported in 1996 on octadecylphosphonic acid self-assembled monolayer (SAM) on mica which was characterized by atomic force microscopy (AFM), confirming its chemical structure.139 The formation of the monolayer was optimized in a three steps process in varying immersion time: (i) nucleation of rounded island, (ii) growth of these micro-domains and then, (iii) coalescence of them to form a homogeneous layer. The same system was monitored by in situ AFM imaging during the film formation.140 The mechanisms of nucleation, growth and coalescence were considered and a kinetic law was found for short time (growth = concentration × time1/2 (mM sec1/2)). A last, the same team studied in 1997 the kinetics of the film formation.141 The monolayer formation was not limited by the adsorption rate of the phosphonic acid onto the surface but by diffusion kinetics of the anchor to the substrate. Unfortunately, no chemical analysis data of the grafting link between phosphonic acid and mica was detailed by the authors. Ten years later, the SAM formation of semi-fluorinated phosphonic acids onto mica was reported. First, clusters of semi-fluorinated nonadecylphosphonic acid (F8H11PO3) were obtained by the Langmuir Blodgett method and then hexadecylphosphonic acid was progressively added.142 This new acid penetrated clusters of semi-fluorinated acid and swelled it since a critical concentration was reached. Beyond this concentration, hexadecylphosphonic acid segregated in a new grafted solid phase. Then, comparison of different molecular monolayers of semifluorinated phosphonic acids (F8H11PO3, F10H6PO3, F8H8PO3, and F6H10PO3) was carried out.143 Continuous SAMs were obtained for phosphonic acids bearing fluorinated part bigger than the hydrocarbonated one. With all other acids, clusters nucleated and grew but did not exhibit coalescence. According to the authors, phase segregation occurred and stopped the SAM formation. The initial acid concentration had no influence on the final repartition and structure on the surface. Similarly to Woodward et al. previous studies, no analysis of the grafted molecules was reported and no data concerning the chemical bonding of phosphonic acid onto the mica substrate were provided. A third team from Brazil published in the same period three papers where the formation of SAMs of octyl and dodecyl phosphonic acid on mica was detailed. They mainly focused on structures formed by these acids after adsorption on the surface. The first paper was centered on the effects of the polarity of the substrate and the temperature on the final SAM structure.144 On mica, the acidic head ensured the contact with substrate and classical multilayers were formed. By increasing temperature, these layers lost their organization around 110 °C. On graphite, alkyl chains interacted with the substrate and formed a bilayer “parallel” to the surface which grew when the temperature increased. In a second publication, quantities of octadecyl and octylphosphonic acid varied and the influence of water on the layer morphology was studied.145 Taking into account these parameters, a lot of different structures (monolayers, bilayers, rods, and branches) were created showing the versatility of the system. In the third study, long range order was monitored by wide-angle scattering in respect to temperature and a phase diagram was extracted.146 Unfortunately, the chemical absorption of phosphonic acid was not detailed. Another contribution reported nanowires of grafted poly(methacryloyloxyethyldimethylbenzylammonium chloride) onto a silicone by moving a droplet of this polymer in solution.147 The formed network structure behaved as a positive template. In a second step, (12-pyrrol-1-yl-dodecyl)phosphonic acid in its anionic form (pH = 11) was complexed with the polyammonium template by the same deposition technique. This procedure led to a network of nanowire composed of grafted ammonium polymer complexed by phosphonic acid. In the last step, this network was transferred on freshly cleaved mica by a printing process (direct contact between the two surfaces). The (12-pyrrol-1-yl-dodecyl)phosphonic acid bore an alkyl chain ended by a pyrrole group which could be polymerized to create conductive rods. In the aim of creating a nano electrical network, the regularity of the deposition technique had to be improved. Once again the chemical grafting on mica was not well characterized in this publication. To sum up this part, interactions of phosphonic acid with mica have been studied during the last two decades but only in the purpose to form self-assembled monolayers. Unfortunately, in all the papers, the interest was more centered on the structure formed than on the study of interaction between phosphonic acid and mica surface.
Concerning montmorillonite, two papers were published dealing with the interactions of glyphosate herbicide with such clay. Glyphosate is a common agricultural product and its behavior in field is an environmental issue. A first paper based on computer simulations studied the different bonding modes of two molecules coming from the degradation of glyphosate in soil.148 The first moiety, sarcosine, seemed to bond mainly by the carboxylic part to the sodium ion contained in the montmorillonite clay. The second degradation product, aminomethylphosphonic acid, interacted with clay though its phosphonic acid. Calculated conformations showed mainly hydrogen bonding but chemical grafting was also suspected. In another paper, authors used XPS and X-ray diffraction (XRD) to confirm the bonding mode.149 Varying the pH, it was shown that the positively charged amino moiety was the principal responsible of adsorption of glyphosate onto montmorillonite. The second way of interaction resulted from phosphonic acid grafting. Mono- and bidentate bonding modes were revealed but no tridentate species was identified.
Finally, last example dealt with the functionalization of natural alumino-silicate nanotubes. The recent developments of nanocomposites based on imogolite were reviewed by a Japanese team.150 Imogolite is composed of natural nanotubes and was discovered on a Japanese volcano in 1961. The way of grafting these nanotubes with phosphonic acid to improve their compatibility with polymers was described.150 The authors first reported the grafting of simple octadecylphosphonic acid permitting a better dispersion of imogolite in polymer but showed a weak interface between polymer and imogolite. This is why a “grafting from” technique was developed (Fig. 4), leading to improved mechanical properties (storage modulus) and permitting to obtain transparent polymethylmethacrylate-based composites.
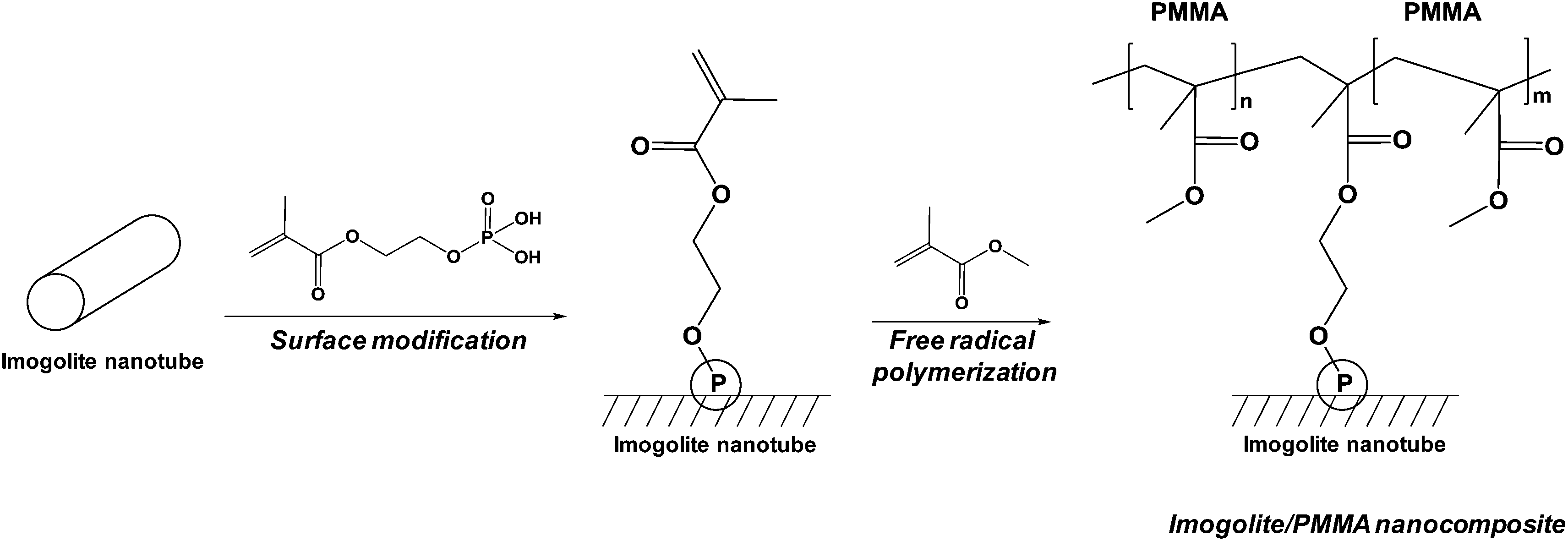 | ||
| Fig. 4 Functionalization of imogolite nanotubes by a phosphonic acid. | ||
A second contribution dealt with the use of halloysite nanotubes modified by phenylphosphonic acid to improve fire properties of Polyamide 6.151 XRD showed a slight expansion of the d-spacing after the treatment with phenylphosphonic acid. After being grafted on halloysite, the phosphonic acid thermal stability was greatly increased as show by thermogravimetric analysis (TGA) measurements. Additionally, energy-dispersive X-ray spectroscopy measurements revealed the presence of phosphorus in nanotubes after treatment. Incorporating 10% of untreated halloysite nanotubes in polyamide improved its fire resistance. The use of modified nanotubes had a greater effect since it divided the peak of heat release by a factor two. This improvement probably came from the formation of a protective char layer between the material and the upper flame.
To conclude, the major part of the literature dealing with phosphonic acids and alumino-silicate was related to the formation of self-assembled layers on cleaved mica but only few details in relation with the structures formed by phosphonic moieties were given. More accurate studies would deserve to be carried out in the future to develop new hybrid materials based on clays, and to better understand the interaction between phosphonic acid derivatives and clays. The hybrid materials obtained could find valuable applications in various fields, for the drug delivery of bioactive molecules, or in the area of protection of polymeric materials against fire, for instance. As a result, in the current trend of “green chemistry”, we assumed that such kind of materials will be developed in the next future.
Notes and references
- R. Francis, N. Joy, E. P. Aparna and R. Vijayan, Polym. Rev., 2014, 54, 268–347 CrossRef CAS.
- K. Farbod, M. R. Nejadnik, J. A. Jansen and S. C. G. Leeuwenburgh, Tissue Eng., Part B, 2014, 20, 173–188 CrossRef CAS PubMed.
- C. Sanchez, C. Boissiere, S. Cassaignon, C. Chaneac, O. Durupthy, M. Faustini, D. Grosso, C. Laberty-Robert, L. Nicole, D. Portehault, F. Ribot, L. Rozes and C. Sassoye, Chem. Mater., 2014, 26, 221–238 CrossRef CAS.
- N. Karsi, P. Lang, M. Chehimi, M. Delamar and G. Horowitz, Langmuir, 2006, 22, 3118–3124 CrossRef CAS PubMed.
- N. Niamsa, C. Kaewtong, W. Srinonmuang, B. Wanno, B. Pulpoka and T. Tuntulani, Polym. Chem., 2013, 4, 3039–3046 RSC.
- A. S. O. Moscofian and C. Airoldi, J. Hazard. Mater., 2008, 160, 63–69 CrossRef CAS PubMed.
- I. Sadaba, M. Ojeda, R. Mariscal and M. L. Granados, Appl. Catal., B, 2014, 150, 421–431 CrossRef PubMed.
- P. H. Mutin, G. Guerrero and A. Vioux, C. R. Chim., 2003, 6, 1153–1164 CrossRef CAS PubMed.
- K. Chougrani, PhD Thesis, Université Montpellier 2, 2007.
- S. Monge and G. David, Phosphorus-based Polymers: From Synthesis to Applications, RSC, 2014, 10.1039/9781782621515.
- G. David, E. Ortega, K. Chougrani, A. Manseri and B. Boutevin, React. Funct. Polym., 2011, 71, 599–606 CrossRef CAS PubMed.
- M. Essahli, G. Colomines, S. Monge, J. J. Robin, A. Collet and B. Boutevin, Polymer, 2008, 49, 4510–4518 CrossRef CAS PubMed.
- H. Vahabi, C. Longuet, L. Ferry, G. David, J. J. Robin and J. M. Lopez-Cuesta, Polym. Int., 2012, 61, 129–134 CrossRef CAS PubMed.
- G. Guerrero, J. G. Alauzun, M. Granier, D. Laurencin and P. H. Mutin, Dalton Trans., 2013, 42, 12569–12585 RSC.
- V. Zoulalian, S. Zuercher, S. Tosatti, M. Textor, S. Monge and J.-J. Robin, Langmuir, 2010, 26, 74–82 CrossRef CAS PubMed.
- P. G. Mingalyov, M. V. Buchnev and G. V. Lisichkin, Russ. Chem. Bull., 2001, 50, 1693–1695 CrossRef CAS.
- M. Eschner, R. Frenzel, F. Simon, D. Pleul, P. Uhlmann and H. Adler, Macromol. Symp., 2004, 210, 77–84 CrossRef CAS PubMed.
- A. G. Koutsioubas, N. Spiliopoulos, D. L. Anastassopoulos, A. A. Vradis and G. D. Priftis, Surf. Interface Anal., 2009, 41, 897–903 CrossRef CAS PubMed.
- I. L. Liakos, E. McAlpine, X. Y. Chen, R. Newman and M. R. Alexander, Appl. Surf. Sci., 2008, 255, 3276–3282 CrossRef CAS PubMed.
- R. Hofer, M. Textor and N. D. Spencer, Langmuir, 2001, 17, 4014–4020 CrossRef CAS.
- R. F. Roskamp, I. K. Vockenroth, N. Eisenmenger, J. Braunagel and I. Koper, ChemPhysChem, 2008, 9, 1920–1924 CrossRef CAS PubMed.
- M. J. Brukman, G. O. Marco, T. D. Dunbar, L. D. Boardman and R. W. Carpick, Langmuir, 2006, 22, 3988–3998 CrossRef CAS PubMed.
- G. Fonder, J. Delhalle, M. Essahli, B. Ameduri and Z. Mekhalif, Surf. Interface Anal., 2008, 40, 85–96 CrossRef CAS PubMed.
- G. Ilia and M. Drehe, Fire Mater., 2010, 34, 271–283 CrossRef CAS PubMed.
- S. Faure, S. Volland, Q. Crouzet, G. Boutevin and C. Loubat, Colloids Surf., A, 2011, 382, 139–144 CrossRef CAS PubMed.
- E. Laiti, L. O. Ohman, J. Nordin and S. Sjoberg, J. Colloid Interface Sci., 1995, 175, 230–238 CrossRef CAS.
- R. M. Stoltenberg, C. Liu and Z. N. Bao, Langmuir, 2011, 27, 445–451 CrossRef CAS PubMed.
- G. Guerrero, P. H. Mutin and A. Vioux, J. Mater. Chem., 2001, 11, 3161–3165 RSC.
- D. Villemin, B. Moreau, F. Simeon, G. Maheut, C. Fernandez, V. Montouillout, V. Caignaert and P. A. Jaffres, Chem. Commun., 2001, 2060–2061 RSC.
- P. H. Mutin, V. Lafond, A. F. Popa, M. Granier, L. Markey and A. Dereux, Chem. Mater., 2004, 16, 5670–5675 CrossRef CAS.
- E. Laiti and L. O. Ohman, J. Colloid Interface Sci., 1996, 183, 441–452 CrossRef CAS.
- N. Tsud and M. Yoshitake, Surf. Sci., 2007, 601, 3060–3066 CrossRef CAS PubMed.
- A. Rumpel, M. Novak, J. Walter, B. Braunschweig, M. Halik and W. Peukert, Langmuir, 2011, 27, 15016–15023 CrossRef CAS PubMed.
- N. Cinausero, N. Azema, M. Cochez, M. Ferriol, M. Essahli, F. Ganachaud and J. M. Lopez-Cuesta, Polym. Adv. Technol., 2008, 19, 701–709 CrossRef CAS PubMed.
- J. J. Shephard, S. A. Dickie and A. J. McQuillan, Langmuir, 2010, 26, 4048–4056 CrossRef CAS PubMed.
- N. Cinausero, N. Azema, J. M. L. Cuesta, M. Cochez and M. Ferriol, Polym. Adv. Technol., 2011, 22, 1931–1939 CrossRef CAS PubMed.
- J. Pahnke and J. Ruhe, Macromol. Rapid Commun., 2004, 25, 1396–1401 CrossRef CAS PubMed.
- W. Gao, L. Dickinson, C. Grozinger, F. G. Morin and L. Reven, Langmuir, 1996, 12, 6429–6435 CrossRef CAS.
- R. Luschtinetz, J. Frenzel, T. Milek and G. Seifert, J. Phys. Chem. C, 2009, 113, 5730–5740 CAS.
- M. Nilsing, S. Lunell, P. Persson and L. Ojamae, Surf. Sci., 2005, 582, 49–60 CrossRef CAS PubMed.
- G. Guerrero, P. H. Mutin and A. Vioux, Chem. Mater., 2001, 13, 4367–4373 CrossRef CAS.
- F. Brodard-Severac, G. Guerrero, J. Maquet, P. Florian, C. Gervais and P. H. Mutin, Chem. Mater., 2008, 20, 5191–5196 CrossRef CAS.
- P. Falaras, I. M. Arabatzis, T. Stergiopoulos, G. Papavassiliou and M. Karagianni, J. Mater. Process. Technol., 2005, 161, 276–281 CrossRef CAS PubMed.
- V. Lafond, C. Gervais, J. Maquet, D. Prochnow, F. Babonneau and P. H. Mutin, Chem. Mater., 2003, 15, 4098–4103 CrossRef CAS.
- E. S. Gawalt, M. J. Avaltroni, N. Koch and J. Schwartz, Langmuir, 2001, 17, 5736–5738 CrossRef CAS.
- S. Pawsey, K. Yach and L. Reven, Langmuir, 2002, 18, 5205–5212 CrossRef CAS.
- R. Helmy and A. Y. Fadeev, Langmuir, 2002, 18, 8924–8928 CrossRef CAS.
- B. Adolphi, E. Jahne, G. Busch and X. D. Cai, Anal. Bioanal. Chem., 2004, 379, 646–652 CrossRef CAS PubMed.
- G. J. Ruiterkamp, M. A. Hempenius, H. Wormeester and G. J. Vancso, J. Nanopart. Res., 2011, 13, 2779–2790 CrossRef CAS.
- B. Barthelemy, S. Devillers, I. Minet, J. Delhalle and Z. Mekhalif, J. Colloid Interface Sci., 2011, 354, 873–879 CrossRef CAS PubMed.
- M. N. Tchoul, S. P. Fillery, H. Koerner, L. F. Drummy, F. T. Oyerokun, P. A. Mirau, M. F. Durstock and R. A. Vaia, Chem. Mater., 2010, 22, 1749–1759 CrossRef CAS.
- S. Oberoi, E. Jaehne and H. J. P. Adler, Macromol. Symp., 2007, 254, 284–291 CrossRef CAS PubMed.
- V. Zoulalian, S. Monge, S. Zuercher, M. Textor, J. J. Robin and S. Tosatti, J. Phys. Chem. B, 2006, 110, 25603–25605 CrossRef CAS PubMed.
- C. Viornery, Y. Chevolot, D. Leonard, B. O. Aronsson, P. Pechy, H. J. Mathieu, P. Descouts and M. Gratzel, Langmuir, 2002, 18, 2582–2589 CrossRef CAS.
- A. Zeller, A. Musyanovych, M. Kappl, A. Ethirajan, M. Dass, D. Markova, M. Klapper and K. Landfester, ACS Appl. Mater. Interfaces, 2010, 2, 2421–2428 CAS.
- I. Rehor, V. Kubicek, J. Kotek, P. Hermann, I. Lukes, J. Szakova, L. V. Elst, R. N. Muller and J. A. Peters, J. Mater. Chem., 2009, 19, 1494–1500 RSC.
- I. Rehor, V. Kubicek, J. Kotek, P. Hermann, J. Szakova and I. Lukes, Eur. J. Inorg. Chem., 2011, 1981–1989 CrossRef CAS PubMed.
- M. Nilsing, P. Persson, S. Lunell and L. Ojamae, J. Phys. Chem. C, 2007, 111, 12116–12123 CAS.
- J. C. Liu, W. L. Wang, H. Z. Yu, Z. L. Wu, J. B. Peng and Y. Cao, Sol. Energy Mater. Sol. Cells, 2008, 92, 1403–1409 CrossRef CAS PubMed.
- U. Lafont, L. Simonin, M. Gaberscek and E. M. Kelder, J. Power Sources, 2007, 174, 1104–1108 CrossRef CAS PubMed.
- W. Gao, L. Dickinson, C. Grozinger, F. G. Morin and L. Reven, Langmuir, 1997, 13, 115–118 CrossRef CAS.
- C. J. Lomoschitz, B. Feichtenschlager, N. Moszner, M. Puchberger, K. Mueller, M. Abele and G. Kickelbick, Langmuir, 2011, 27, 3534–3540 CrossRef CAS PubMed.
- B. Feichtenschlager, C. J. Lornoschitz and G. Kickelbick, J. Colloid Interface Sci., 2011, 360, 15–25 CrossRef CAS PubMed.
- D. Carriere, M. Moreau, P. Barboux, J. P. Boilot and O. Spalla, Langmuir, 2004, 20, 3449–3455 CrossRef CAS.
- S. Pawsey, M. McCormick, S. De Paul, R. Graf, Y. S. Lee, L. Reven and H. W. Spiess, J. Am. Chem. Soc., 2003, 125, 4174–4184 CrossRef CAS PubMed.
- S. Marcinko and A. Y. Fadeev, Langmuir, 2004, 20, 2270–2273 CrossRef CAS.
- J. Randon, P. Blanc and R. Paterson, J. Membr. Sci., 1995, 98, 119–129 CrossRef CAS.
- A. M. Clausen and P. W. Carr, Anal. Chem., 1998, 70, 378–385 CrossRef CAS.
- M. Sajjad, B. Feichtenschlager, S. Pabisch, J. Svehla, T. Koch, S. Seidler, H. Peterlik and G. Kickelbick, Polym. Int., 2012, 61, 274–285 CrossRef CAS PubMed.
- Y.-g. Ha, J. D. Emery, M. J. Bedzyk, H. Usta, A. Facchetti and T. J. Marks, J. Am. Chem. Soc., 2011, 133, 10239–10250 CrossRef CAS PubMed.
- A. B. Kashyout, H. M. A. Soliman, H. S. Hassan and A. M. Abousehly, J. Nanomater., 2010, 341841 Search PubMed.
- T. Wang, H. Wu, Z. Wang, C. Chen and C. Liu, Nanoscale Res. Lett., 2013, 8, 1–5 CrossRef PubMed.
- M. Baum, S. Polster, M. P. M. Jank, I. Alexeev, L. Frey and M. Schmidt, Appl. Phys. A: Mater. Sci. Process., 2012, 107, 269–273 CrossRef CAS.
- N. A. Estrich, D. H. Hook, A. N. Smith, J. T. Leonard, B. Laughlin and J. P. Maria, J. Appl. Phys., 2013, 113 Search PubMed.
- L. C. Chao, C. C. Ye, Y. P. Chen and H. Z. Yu, Appl. Surf. Sci., 2013, 282, 384–389 CrossRef CAS PubMed.
- A. J. Tzou, K. F. Chien, H. Y. Lai, J. T. Ku, L. Lee, W. C. Fan and W. C. Chou, J. Cryst. Growth, 2013, 378, 466–469 CrossRef CAS PubMed.
- H. Z. Zhuang, P. Xu and J. L. Li, Spectrochim. Acta, Part A, 2013, 110, 395–399 CrossRef CAS PubMed.
- G. C. Fan, J. Chen, Y. J. Ma and Z. Y. Huang, Micro Nano Lett., 2012, 7, 795–797 Search PubMed.
- H. T. Lin, Y. S. Wu, X. Q. Cao and H. B. Fu, J. Phys. Chem. C, 2012, 116, 21657–21663 CAS.
- D. Liu, W. Wu, Y. Qiu, S. Yang, S. Xiao, Q. Q. Wang, L. Ding and J. Wang, Langmuir, 2008, 24, 5052–5059 CrossRef CAS PubMed.
- C. Bressy, V. G. Ngo, F. Ziarelli and A. Margaillan, Langmuir, 2012, 28, 3290–3297 CrossRef CAS PubMed.
- E. Smecca, A. Motta, M. E. Fragala, Y. Aleeva and G. G. Condorelli, J. Phys. Chem. C, 2013, 117, 5364–5372 CAS.
- X. Q. Shi, H. Xu, M. A. Van Hove, N. H. Moreira, A. L. Rosa and T. Frauenheim, Surf. Sci., 2012, 606, 289–292 CrossRef CAS PubMed.
- P. J. Hotchkiss, M. Malicki, A. J. Giordano, N. R. Armstrong and S. R. Marder, J. Mater. Chem., 2011, 21, 3107–3112 RSC.
- C. L. Perkins, J. Phys. Chem. C, 2009, 113, 18276–18286 CAS.
- T. Stubhan, M. Salinas, A. Ebel, F. C. Krebs, A. Hirsch, M. Halik and C. J. Brabec, Adv. Energy Mater., 2012, 2, 532–535 CrossRef CAS PubMed.
- K. Beltsios, P. Normand, E. Kapetanakis, D. Tsoukalas and A. Travlos, Microelectron. Eng., 2002, 61–2, 631–635 CrossRef.
- R. R. Rye, G. C. Nelson and M. T. Dugger, Langmuir, 1997, 13, 2965–2972 CrossRef CAS.
- E. L. Hanson, J. Schwartz, B. Nickel, N. Koch and M. F. Danisman, J. Am. Chem. Soc., 2003, 125, 16074–16080 CrossRef CAS PubMed.
- J. B. Miller, J. Schwartz and S. L. Bernasek, J. Am. Chem. Soc., 1993, 115, 8239–8247 CrossRef CAS.
- J. L. Vivero-Escoto, M. Carboni, C. W. Abney, K. E. deKrafft and W. B. Lin, Microporous Mesoporous Mater., 2013, 180, 22–31 CrossRef CAS PubMed.
- X. L. Wang, L. Y. Yuan, Y. F. Wang, Z. J. Li, J. H. Lan, Y. L. Liu, Y. X. Feng, Y. L. Zhao, Z. F. Chai and W. Q. Shi, Sci. China: Chem., 2012, 55, 1705–1711 CrossRef CAS.
- N. Rozlosnik, M. C. Gerstenberg and N. B. Larsen, Langmuir, 2003, 19, 1182–1188 CrossRef CAS.
- P. Thissen, A. Vega, T. Peixoto and Y. J. Chabal, Langmuir, 2012, 28, 17494–17505 CrossRef CAS PubMed.
- S. Lassiaz, D. Labarre, A. Galarneau, D. Brunel and P. H. Mutin, J. Mater. Chem., 2011, 21, 8199–8205 RSC.
- O. Acton, D. Hutchins, L. Arnadottir, T. Weidner, N. Cernetic, G. G. Ting, T. W. Kim, D. G. Castner, H. Ma and A. K. Y. Jen, Adv. Mater., 2011, 23, 1899–1902 CrossRef CAS PubMed.
- C. W. Hsu, H. R. Liou, W. F. Su and L. Y. Wang, J. Colloid Interface Sci., 2008, 324, 236–239 CrossRef CAS PubMed.
- A. Vega, P. Thissen and Y. J. Chabal, Langmuir, 2012, 28, 8046–8051 CrossRef CAS PubMed.
- M. Dubey, T. Weidner, L. J. Gamble and D. G. Castner, Langmuir, 2010, 26, 14747–14754 CrossRef CAS PubMed.
- D. Angmo and F. C. Krebs, J. Appl. Polym. Sci., 2013, 129, 1–14 CrossRef CAS PubMed.
- X. Chen, E. Luais, N. Darwish, S. Ciampi, P. Thordarson and J. J. Gooding, Langmuir, 2012, 28, 9487–9495 CrossRef CAS PubMed.
- S. Besbes, A. Ltaief, K. Reybier, L. Ponsonnet, N. Jaffrezic, J. Davenas and H. Ben Ouada, Synth. Met., 2003, 138, 197–200 CrossRef CAS.
- C. J. Liu, K. Oshima, M. Shimomura and S. Miyauchi, J. Appl. Polym. Sci., 2006, 100, 1881–1888 CrossRef CAS PubMed.
- S. H. Jee, S. H. Kim, J. H. Ko, D. J. Kim and Y. S. Yoon, J. Ceram. Process. Res., 2008, 9, 42–45 Search PubMed.
- R. Schlapak, D. Armitage, N. Saucedo-Zeni, M. Hohage and S. Howorka, Langmuir, 2007, 23, 10244–10253 CrossRef CAS PubMed.
- P. J. Hotchkiss, S. C. Jones, S. A. Paniagua, A. Sharma, B. Kippelen, N. R. Armstrong and S. R. Marder, Acc. Chem. Res., 2012, 45, 337–346 CrossRef CAS PubMed.
- B. Zou, X. J. Zhang, Y. Wang, C. Gong, Y. P. Zhang, J. S. Jie, W. Deng and X. H. Zhang, New J. Chem., 2013, 37, 1776–1781 RSC.
- B. Vercelli, G. Zotti, G. Schiavon, S. Zecchin and A. Berlin, Langmuir, 2003, 19, 9351–9356 CrossRef CAS.
- M. D. Losego, J. T. Guske, A. Efremenko, J. P. Maria and S. Franzen, Langmuir, 2011, 27, 11883–11888 CrossRef CAS PubMed.
- J. A. Bardecker, H. Ma, T. Kim, F. Huang, M. S. Liu, Y. J. Cheng, G. Ting and A. K. Y. Jen, Adv. Funct. Mater., 2008, 18, 3964–3971 CrossRef CAS PubMed.
- S. Besbes, H. Ben Ouada, J. Davenas, L. Ponsonnet, N. Jaffrezic and P. Alcouffe, Mater. Sci. Eng., C, 2006, 26, 505–510 CrossRef CAS PubMed.
- N. Doubina, J. L. Jenkins, S. A. Paniagua, K. A. Mazzio, G. A. MacDonald, A. K. Y. Jen, N. R. Armstrong, S. R. Marder and C. K. Luscombe, Langmuir, 2012, 28, 1900–1908 CrossRef CAS PubMed.
- A. Sharma, B. Kippelen, P. J. Hotchkiss and S. R. Marder, Appl. Phys. Lett., 2008, 93, 163308 CrossRef PubMed.
- M. Q. Wang and I. G. Hill, Org. Electron., 2012, 13, 498–505 CrossRef CAS PubMed.
- J. Guo, N. Koch, S. L. Bernasek and J. Schwartz, Chem. Phys. Lett., 2006, 426, 370–373 CrossRef CAS PubMed.
- X. Chen, M. Chockalingam, G. Z. Liu, E. Luais, A. L. Gui and J. J. Gooding, Electroanalysis, 2011, 23, 2633–2642 CrossRef CAS PubMed.
- S. A. Paniagua, P. J. Hotchkiss, S. C. Jones, S. R. Marder, A. Mudalige, F. S. Marrikar, J. E. Pemberton and N. R. Armstrong, J. Phys. Chem. C, 2008, 112, 7809–7817 CAS.
- N. W. Polaske, H. C. Lin, A. Tang, M. Mayukh, L. E. Oquendo, J. T. Green, E. L. Ratcliff, N. R. Armstrong, S. S. Saavedra and D. V. McGrath, Langmuir, 2011, 27, 14900–14909 CrossRef CAS PubMed.
- T. Kasahara, S. Matsunami, T. Edura, J. Oshima, C. Adachi, S. Shoji and J. Mizuno, Sens. Actuators, A, 2013, 195, 219–223 CrossRef CAS PubMed.
- S. H. Kim, S. J. Baek, Y. C. Chang and H. J. Chang, Phys. Status Solidi A, 2012, 209, 2317–2323 CrossRef CAS PubMed.
- B. J. Leever, I. P. Murray, M. F. Durstock, T. J. Marks and M. C. Hersam, J. Phys. Chem. C, 2011, 115, 22688–22694 CAS.
- M. Takemoto, S. Fujibayashi, M. Neo, J. Suzuki, T. Matsushita, T. Kokubo and T. Nakamura, Biomaterials, 2006, 27, 2682–2691 CrossRef CAS PubMed.
- A. Altin, B. Akgun, O. Buyukgumus, Z. S. Bilgici, S. Agopcan, D. Asik, H. Y. Acar and D. Avci, React. Funct. Polym., 2013, 73, 1319–1326 CrossRef CAS PubMed.
- A. Altin, B. Akgun, Z. S. Bilgici, S. B. Turker and D. Avci, J. Polym. Sci., Part A: Polym. Chem., 2014, 52, 511–522 CrossRef CAS PubMed.
- B. Canniccioni, S. Monge, G. David and J.-J. Robin, Polym. Chem., 2013, 4, 3676–3685 RSC.
- V. Besse, L. Le Pluart, W. D. Cook, T. N. Pham and P. J. Madec, J. Polym. Sci., Part A: Polym. Chem., 2013, 51, 149–157 CrossRef CAS PubMed.
- G. David, C. Negrell-Guirao, F. Iftene, B. Boutevin and K. Chougrani, Polym. Chem., 2012, 3, 265–274 RSC.
- N. Moszner, J. Angermann, U. Fischer and T. Bock, Macromol. Mater. Eng., 2013, 298, 454–461 CrossRef CAS PubMed.
- G. Sahin, D. Avci, O. Karahan and N. Moszner, J. Appl. Polym. Sci., 2009, 114, 97–106 CrossRef CAS PubMed.
- N. Sibold, P. J. Madec, S. Masson and T. N. Pham, Polymer, 2002, 43, 7257–7267 CrossRef CAS.
- F. Zeuner, J. Angermann and N. Moszner, Synth. Commun., 2006, 36, 3679–3691 CrossRef CAS.
- Y. Yoshida, B. Van Meerbeek, Y. Nakayama, M. Yoshioka, J. Snauwaert, Y. Abe, P. Lambrechts, G. Vanherle and M. Okazaki, J. Dent. Res., 2001, 80, 1565–1569 CrossRef CAS PubMed.
- M. A. Bayle, G. Gregoire and P. Sharrock, J. Dent., 2007, 35, 302–308 CrossRef CAS PubMed.
- M. A. Bayle, K. Nasr, G. Gregoire and P. Sharrock, Dent. Mater., 2008, 24, 386–391 CrossRef CAS PubMed.
- B. Zhang, L. Zhang, F. Li, W. Hu and P. M. Hannam, Corros. Sci., 2010, 52, 3883–3890 CrossRef CAS PubMed.
- M. Sedlak and H. Colfen, Macromol. Chem. Phys., 2001, 202, 587–597 CrossRef CAS.
- A. T. Kan, G. M. Fu and M. B. Tomson, J. Colloid Interface Sci., 2005, 281, 275–284 CrossRef CAS PubMed.
- W. El Malti, D. Laurencin, G. Guerrero, M. E. Smith and P. H. Mutin, J. Mater. Chem., 2012, 22, 1212–1218 RSC.
- J. T. Woodward, A. Ulman and D. K. Schwartz, Langmuir, 1996, 12, 3626–3629 CrossRef CAS.
- J. T. Woodward and D. K. Schwartz, J. Am. Chem. Soc., 1996, 118, 7861–7862 CrossRef CAS.
- J. T. Woodward, I. Doudevski, H. D. Sikes and D. K. Schwartz, J. Phys. Chem. B, 1997, 101, 7535–7541 CrossRef CAS.
- S. Trabelsi, S. Zhang, T. R. Lee and D. K. Schwartz, Soft Matter, 2007, 3, 1518–1524 RSC.
- S. Trabelsi, S. Zhang, Z. Zhang, T. R. Lee and D. K. Schwartz, Soft Matter, 2009, 5, 750–758 RSC.
- G. N. Fontes and B. R. A. Neves, Langmuir, 2005, 21, 11113–11118 CrossRef CAS PubMed.
- M. C. Prado and B. R. A. Neves, Langmuir, 2010, 26, 648–654 CrossRef CAS PubMed.
- M. de Pauli, M. d. C. Prado, M. J. Souza Matos, G. N. Fontes, C. A. Perez, M. S. Carvalho Mazzoni, B. R. Almeida Neves and A. Malachias, Langmuir, 2012, 28, 15124–15133 CrossRef CAS PubMed.
- K. Demidenok, V. Bocharova, M. Stamm, E. Jaehne, H.-J. P. Adler and A. Kiriy, Langmuir, 2007, 23, 9287–9292 CrossRef CAS PubMed.
- A. Rennig, A. Slutter and L. Tribe, Int. J. Quantum Chem., 2008, 108, 538–543 CrossRef CAS PubMed.
- G. A. Khoury, T. C. Gehris, L. Tribe, R. M. T. Sanchez and M. D. Afonso, Appl. Clay Sci., 2010, 50, 167–175 CrossRef CAS PubMed.
- W. O. Yah, K. Yamamoto, N. Jiravanichanun, H. Otsuka and A. Takahara, Materials, 2010, 3, 1709–1745 CrossRef CAS PubMed.
- D. C. O. Marney, W. Yang, L. J. Russell, S. Z. Shen, T. Nguyen, Q. Yuan, R. Varley and S. Li, Polym. Adv. Technol., 2012, 23, 1564–1571 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |