
Role of zinc and copper metal ions in amyloid β-peptides Aβ1–40 and Aβ1–42 aggregation†
Abstract
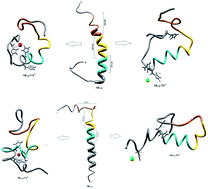
The aggregation of amyloid β-peptides (Aβ) plays a key role in the central pathological pathways in Alzheimer's disease. The interaction of zinc and copper metal ions with Aβ-peptide gives considerable understanding to fight this incurable disease. The different conformations of native and metal interacting Aβ-peptides were investigated using molecular dynamics simulations for 50 ns. Aβ1–40–Zn2+ peptide adopts a β-hairpin structure, which was promoted by the turn region (Val24-Asp27) and increases the hydrophobic contact between the central hydrophobic (CHC) (Leu17-Ala21) and c-terminal (Met35-Val40). The turn region was stabilized by forming a salt-bridge between Asp23-Lys28 and Glu22-Lys28 residues. The structure of the Aβ1–42–Zn2+ peptide was destabilized by the β-hairpin structure due to the absence of the salt-bridge, and reduces the hydrophobic contact between CHC and the c-terminal, which results in Aβ1–42 monomer disaggregation. The Aβ1–40–Cu2+ peptide has three bend regions, which are separated by a coil segment. These segments reduce the hydrophobic contact between CHC and the c-terminal, and promote the formation of a salt-bridge which in turn destabilizes the turn region of the peptide. The hairpin conformation of the Aβ1–42–Cu2+ structure was stabilized by increasing the hydrophobic contact and the salt-bridge which is facilitated for aggregation.
- This article is part of the themed collection: Towards understanding and treating Alzheimer’s disease
 Please wait while we load your content...
Please wait while we load your content...

