
Hybrid periodic mesoporous organosilica designed to improve the properties of immobilized enzymes†
Abstract
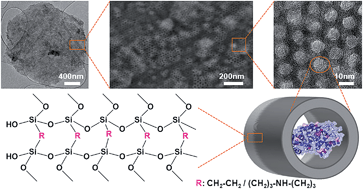
Two types of highly ordered periodic mesoporous organosilicas have been synthesized as tailor-made supports to immobilize two different enzymes, lipase and laccase. These materials provide an environment where abundant organic groups in close vicinity to the enzyme surface generate a high chemical affinity, which results in high values of enzyme loading, catalytic activity and stabilization. A hydrophobic periodic mesoporous organosilica support (PMO) with a highly ordered hexagonal arrangement of parallel pore channels with a diameter around 7 nm, containing framework hydrocarbon groups (ethylene), was used for lipase immobilization. A novel periodic mesoporous aminosilica (PMA), containing secondary amine groups in its framework and having expanded pores was synthesized and studied for immobilization of a larger enzyme, namely laccase. The synthesis conditions were adjusted, using bis[(3-trimethoxysilyl)propyl]amine and 1,2-bis(trimethoxysilyl)ethane as framework co-precursors and 1,3,5-triisopropylbenzene as micelle expander for producing large pores (>10 nm). The properties of this multifunctional PMA having hydrophilic amino groups and hydrophobic ethylene/propylene groups within the framework were studied. This work compares the confinement of lipase and laccase enzymes in the pores of these hybrid organosilica materials and its effect on immobilization and stabilization parameters. Laccase immobilized on PMA and lipase immobilized on PMO exhibited higher stability in solvents (ethanol and methanol, respectively) compared to enzymes supported on functionalized silica materials with pending organic groups on the surface. High retention of enzymes inside the pores of these materials has been achieved and leaching has been fully prevented. These results can be attributed to the different interactions (hydrophobic, electrostatic and hydrogen bonding) established between the surfaces of the enzyme and the PMO/PMA support, which are enhanced by an optimum pore size adjusted to the enzyme dimensions.
- This article is part of the themed collection: Porous Materials (FEZA 2014)
 Please wait while we load your content...
Please wait while we load your content...

