
Synthesis of randomly aminated polyvinylpyrrolidone and its use in the preparation of hydrolyzable conjugates†
Abstract
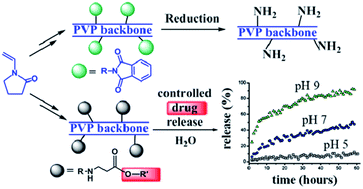
In this work, a versatile synthetic route to functionalize vinylpyrrolidone (VP) with protected or unprotected aliphatic primary amine groups is described for the first time. Using these monomeric precursors polyvinylpyrrolidones (PVPs) with controlled load of side amine groups randomly distributed along the chains have been prepared. These functionalized VP and PVP systems are active molecules highly desirable for further couplings, i.e. they can easily afford the simple preparation of water soluble covalent side chain conjugates onto the PVP backbone. To show the potential of this functionalization, we have prepared functionalized polymers as a controlled release delivering vehicle, employing the bactericide and preservative 2-phenoxyethanol as a hydroxyl-containing model drug.
 Please wait while we load your content...
Please wait while we load your content...

