DOI:
10.1039/C4RA05247A
(Paper)
RSC Adv., 2014,
4, 36940-36950
Synthesis, structural characterization and photocatalytic application of ZnO@ZnS core–shell nanoparticles
Received
2nd June 2014
, Accepted 4th August 2014
First published on 4th August 2014
Abstract
ZnO nanoparticles were synthesized by co-precipitation with no capping agent followed by covering with ZnS using a solution-based chemical method at low temperature. By variation of the solution concentrations it was found that the fully-covering ZnS shell forms by a reaction of Na2S with ZnO NPs followed by the formation of ZnS nano-crystals by the reaction of Na2S with ZnCl2. The mechanism that led to full coverage of the ZnO core is proposed to be the addition of ZnCl2 at a later stage of the growth which guarantees a continuous supply of Zn ions to the core surface. Moreover, the ZnS nano-crystals that uniformly cover the ZnO NPs show no epitaxial relationship between the ZnO core and ZnS shell. The slow atomic mobility at the low reaction temperature is attributed to the non-epitaxial uniform ZnS shell growth. The rough surface of the ZnO grains provides initial nucleation positions for the growth of the ZnS shell nano-crystals. The low growth temperature also inhibits the abnormal growth of ZnS grains and results in the homogeneous coverage of ZnS nano-crystals on the ZnO core surface. The as-synthesized ZnO@ZnS core–shell nanoparticles were used as a photocatalyst to decompose Rose Bengal dye at three different pH values. ZnO@ZnS core–shell nanoparticles perform as a more active photocatalyst at a pH of 4, while pure ZnO nanoparticles are more efficient at a pH of 7.
1. Introduction
Zinc oxide (ZnO) nanoparticles (NPs) as a cheap nontoxic semiconductor with a wide direct band gap (3.37 eV) are a promising material for different applications such as photocatalysts,1 photodetectors,2 gas sensors,3 piezoelectric sensors4 and ultraviolet lasers5 and can be synthesized in different ways such as by the chemical vapor phase method,6 thermal evaporation,7 the vapor-solid technique,8 the hydrothermal method9 and the co-precipitation method.10 Recently, it was found that surface coating with different semiconductors can dramatically change the properties of ZnO nano-crystallites. In recent years, much effort has been devoted to the development of core–shell structured materials on the nanometer scale. The shell usually acts as a barrier between the core and the environment and can alter the charge, functionality, and reactivity of the surface and also it can change the stability and dispersive ability of the core material. Furthermore, electrical, catalytic, optical, or magnetic properties of the core material can be improved by covering with another material. In general, the synthesis of core–shell structured material leads to obtain a new composite material having properties between the core and shell materials.11–13 Zinc sulfide (ZnS) with wide band gap (3.72 eV)14 is one of the candidates for the coating materials and has potential applications as electroluminescent devices,15 sensors and lasers.16 ZnO@ZnS core–shell nanoparticles (CSNPs) are demonstrated improved physical and chemical properties for different applications. Therefore, considerable effort has been devoted to the design and fabrication control of ZnO@ZnS CSNPs. Various ZnO@ZnS core–shell nanostructures have been synthesized, most of them at elevated temperature.17–23 Epitaxial growth of ZnS shell on ZnO core is obtained by the high-temperature syntheses. In contrast, low-temperature synthesis with low cost can lead to a uniform nanocrystalline ZnS shell structure, but so far only a limited number of publications can be found.24,25 There is a lack of simple and fast reliable way for the growth of pure ZnO@ZnS CSNPs with full coverage of the shell material. In the work reported by Nam et al.24 the starting materials and the method are different from the present manuscript, and the grown ZnO@ZnS CSNPs are quite larger in size (more than 200 nm) compared to the core–shell nanoparticles presented in this work. Sharma et al.25 reported a preparation method of the ZnO@ZnS CSNPs using the same precursors as ours, while their HRTEM results show a non-uniform core–shell NPs. Liu et al. successfully obtained a full coverage of ZnS shell at low temperature, but with a more time consuming process than presented herein.26
Today's global problem of water pollution is cause to a large extent by dyeing, which is mainly due to printing industries that discharge their effluent into the natural water resources without any treatment.27–29 Different methods and materials have been developed alleviate this problem. TiO2 has been used as a photocatalyst to degrade different dyes and pollutants, but further investigation has shown that ZnO not only has similar efficiency of photocatalytic degradation but also it is a better substitute for TiO2 in some applications.30–32 Since covering ZnO with different semiconductors changes the electronic states and the surface of the ZnO NPs, this could affect its photocatalytic activity.33 The other issue that needs to be considered is related to the photocatalytic activity of NPs at different pH values. As dyes pollutants can be found in different mediums, investigating the photocatalytic efficiency of products at different pH values can provide useful information.
In this work, we show a simple fast reliable method to grow ZnO@ZnS CSNPs with full coverage using a simple two steps low temperature chemical method. In this way the ZnO NPs are covered with ZnS without using any additional capping agent between them. The products were prepared with different concentration of the sulfur source and were characterized by X-ray diffraction (XRD), Fourier transform infrared spectroscopy (FTIR), scanning electron microscopy (SEM), high-resolution transmission electron microscopy (HRTEM), and energy dispersive X-ray spectroscopy (EDX) mapping. Finally, the as-synthesized ZnO@ZnS CSNPs were successfully used as a photocatalyst to degrade Rose Bengal at different pH values.
2. Experiments
2.1 Materials
Zinc acetate dehydrates (ZnAC2·2H2O) and sodium hydroxide NaOH were used as the starting materials for preparing the ZnO nanoparticles. Sodium sulfide (Na2S), zinc chloride (ZnCl2) and isopropanol were used as precursors for the formation of ZnS over ZnO nanoparticles. All the materials were purchased from Sigma Aldrich and used without any further purification.
2.2 Preparation of ZnO nanoparticles
The zinc oxide nanoparticles were prepared using the co-precipitation method. In this way, zinc acetate dehydrate (ZnAC2·2H2O) and sodium hydroxide NaOH were dissolved in deionized water to form two transparent solutions with 0.5 M and 1 M concentrations, respectively. Then, by two pipettes, these solutions were poured into a beaker at room temperature. The mixture was stirred for 2 h during which a white precipitate was formed in the solution. Then the obtained precipitation was separated by centrifugation. Finally, ZnO nanoparticles were obtained by washing with deionized water and acetone and drying at 75 °C.
2.3 Covering ZnO nanoparticles with ZnS
ZnO nanoparticles were covered with the ZnS using a simple chemical method. 0.3 g of the as-grown ZnO nanoparticles with 50 mL isopropanol was sonicated for 10 min. After adjusting the pH to 10 using ammonium hydroxide, a solution of Na2S was added to the mixture drop wise. Then it was kept under continuous stirring at 60 °C for 2 h. Finally, in order to achieve the formation of ZnO@ZnS CSNPs, a solution of 0.05 M ZnCl2 was added drop wise into the above mixed solution and stirred for 1 h. Afterwards, the product was washed with deionized water and acetone and was dried at 70 °C. In order to investigate the effect of the shell thickness on the structural properties of ZnO@ZnS CSNPs, the samples were synthesized with three concentrations of the Na2S solution (0.025 M, 0.05 M, and 0.1 M).
The structural characterization of ZnO@ZnS CSNPs was investigated by X-ray powder diffraction (XRD) using a Phillips PW 1729 powder diffractometer equipped with CuKα radiation (λ = 1.5418 Å) operating at a generator voltage of 40 kV and a current of 40 mA. A LEO 1550 Gemini field emission scanning electron microscope working at 15 kV was used to investigate the morphology of the ZnO@ZnS CSNPs. An Equinox 55 FT-IR spectrometer was used in order to reach further information about the products and their surfaces. The HRTEM characterization was carried out by using a FEI Tecnai G2 TF20 UT instrument with a field-emission gun operated at 200 kV. The instrument has a point resolution of 0.19 nm and is equipped with an EDX system. The TEM specimen was prepared by dispersing the nanostructure powder on a copper grid with a thin amorphous carbon film. A Perkin Elmer Lambda 900 UV-visible spectrophotometer was used to investigate the photocatalytic activities of the as-synthesized ZnO@ZnS CSNPs.
A home-made photo reactor equipped with four 18 W UV lamps (256 nm) was used to perform photocatalytic experiments. 0.05 g of each sample material was used as photocatalyst after mixing with 100 mL of the aqueous Rose Bengal dye solution with an initial concentration of 40 mg L−1. The dye solutions were prepared with different pH values using three various buffers (4, 7, and 10) in order to investigate the rate of dye removal at different pH values. The solution was put on stirring for 30 min until adsorption equilibrium was achieved then the solution was kept in the reactor and exposed to the UV light. In order to determine the dye concentration using UV-Vis spectrophotometer, some irradiated solution was withdrawn from the beaker each 20 min and their suspended samples were collected by centrifugation.
3. Results and discussion
3.1 Characterizations of the ZnO@ZnS CSNPs
Powder X-ray diffraction was used to determine the structural properties of the products. The XRD patterns of pure ZnO NPs and ZnO@ZnS CSNPs with different concentration of the sulfur source are shown in Fig. 1. For ZnO NPs, the peaks can be indexed to the known the hexagonal structure of ZnO with lattice constants of a = b = 3.254 Å and c = 5.212 Å with an estimated standard deviation of 0.004 Å and 0.005 Å, respectively (JCPDF: 79-2205). The sample is phase-pure as no peaks from other ZnO phases or impurities were observed, let alone a possible amorphous fraction not resolvable in TEM. It is clear that the intensity of the peaks belonging to the ZnO NPs in the diffraction pattern of ZnO@ZnS nanoparticles were lower than pure ZnO NPs and also the more concentration of sulfur source, the less peak intensities. Decreasing the peak intensities can be a sign of increased covering of the ZnO NPs with another material. A careful perusal of this image shows that, for ZnO@ZnS CSNPs there is a small wide peak around 29°, which is getting higher by increasing the concentration of the sulfur source. A comparison with the standard card demonstrates that this peak belongs to ZnS cubic zinc blende (JCPDF: 005-0566) and it is an evidence of the covering the ZnO NPs with ZnS. Comparing this with the XRD results of the work reported by Sharma et al.25 it is clear that the grown ZnO@ZnS CSNPs in this work are pure core–shell NPs and no impurity phases can be observed.
 |
| | Fig. 1 XRD spectra of ZnO NPs and ZnO@ZnS CSNPs. | |
The crystalline size of the ZnO NPs was determined using the Scherrer formula:
where
D is the crystalline size,
λ is the X-ray wavelength,
θ is the Bragg diffraction angle of the peak, and
β is the full-width at half maximum of the diffraction peak. The crystallite size of the ZnO NPs was calculated to be 19 nm.
ATR-FTIR spectroscopy was used to identify the capping of the ZnO NPs by the ZnS. Fig. 2 shows the FTIR spectra of the different samples. The peak which is observed from 400 cm−1 to 500 cm−1 is due to Zn–O vibrations of ZnO NPs. The peak at around 1100 cm−1 is the characteristic ZnS vibration peak that cannot be observed in the pure ZnO and this confirms that the ZnO nanoparticles capped with ZnS. This peak is absent from the pure ZnO NPs spectrum. Also as it can be seen in Fig. 2, the peak around 1100 cm−1 is increased by increasing the concentration of the sulfur source, and that indicates the presence of ZnS covering the ZnO NPs. The peak from 1500 cm−1 to 1650 cm−1 belongs to vibrations of H–O–H bond and the broad peak from 3200 cm−1 to 3650 cm−1 is due to the O–H variation of water molecules.
 |
| | Fig. 2 ATR-FTIR spectra of ZnO NPs and ZnO@ZnS CSNPs. | |
SEM imaging was carried out to determine the morphology of the pure ZnO NPs and to identify possible changes by covering. Fig. 3 shows the SEM images of ZnO NPs and the different ZnO@ZnS CSNPs. It can clearly be seen that, particles are not entirely spherical in shape and the agglomeration of ZnS-capped ZnO NPs is increased with increasing the concentration of the sulfur source. One can imagine that, each ZnO@ZnS CSNP includes a cluster of ZnO NPs which is covered with capping agent (ZnS) and it causes the increase in the size of the ZnO@ZnS CSNP compared to the pure ZnO NPs.
 |
| | Fig. 3 FESEM images of ZnO NPs and ZnO@ZnS CSNPs. | |
HRTEM is the most reliable technique to investigate whether or not the ZnO particles are covered with ZnS and for this reason a closed study was performed on the ZnO@ZnS CSNPs prepared with 0.05 M concentration of Na2S. The TEM image in Fig. 4a shows the grain of ZnO@ZnS CSNPs. The dimension of the ZnO core is approximately 200 nm long and 100 nm wide. The BF-TEM image also shows that the surface of the ZnO core is rather rough. For comparison, pure initial ZnO NPs were characterized as illustrated in Fig. 5a and the STEM of ZnO@ZnS CSNPs is also shown in Fig. 5b. The rough surface of ZnO is beneficial for the reaction of ZnO with Na2S and nucleation of ZnS. To characterize the crystallinity of ZnO NPs, the TEM sample was tilted and a selected area electron diffraction (SAED) pattern along the [11![[2 with combining macron]](https://www.rsc.org/images/entities/char_0032_0304.gif) 0] zone axis of ZnO was obtained (see Fig. 4b). Furthermore, the ZnS shell structure was investigated by energy dispersive X-ray spectroscopy (EDX) mapping. Atomic concentration of Zn is the same for both ZnO and ZnS phases. Thus, to distinguish the ZnO from the ZnS, the distribution of sulfur and oxygen is critical. Fig. 4c exhibits that sulfur distributes on surface (shell), whereas oxygen is in the core region. Furthermore, the HRTEM image from the left corner of the ZnO@ZnS CSNPs is acquired and shown in Fig. 4d. Both the HRTEM image and the corresponding fast Fourier transform (FFT) pattern (Fig. 4e) show that the ZnS is polycrystalline. ZnO has hexagonal wurtzite structure and ZnS has cubic zinc blende structure and ZnS has a larger cell parameters. The circles in Fig. 4b and e indicate the reflections from polycrystalline ZnS phase. No epitaxial relationship between the ZnS and the ZnO was found. Epitaxial relationship between ZnO and ZnS was frequently found in some high temperature synthesized ZnO@ZnS samples18–21 in spite of a large mismatch between ZnO and ZnS structures. In this work, ZnS NPs are uniformly and randomly distributed on ZnO core surface because of the low atomic mobility at low temperature. Fig. 6 shows EDX mapping of the ZnO@ZnS CSNPs prepared with 0.05 M concentration of Na2S. It is clear that increasing the concentration of the S2− ions leads to formation of the more uniform and dense shell. Also, no ZnS NPs was observed in this concentration.
0] zone axis of ZnO was obtained (see Fig. 4b). Furthermore, the ZnS shell structure was investigated by energy dispersive X-ray spectroscopy (EDX) mapping. Atomic concentration of Zn is the same for both ZnO and ZnS phases. Thus, to distinguish the ZnO from the ZnS, the distribution of sulfur and oxygen is critical. Fig. 4c exhibits that sulfur distributes on surface (shell), whereas oxygen is in the core region. Furthermore, the HRTEM image from the left corner of the ZnO@ZnS CSNPs is acquired and shown in Fig. 4d. Both the HRTEM image and the corresponding fast Fourier transform (FFT) pattern (Fig. 4e) show that the ZnS is polycrystalline. ZnO has hexagonal wurtzite structure and ZnS has cubic zinc blende structure and ZnS has a larger cell parameters. The circles in Fig. 4b and e indicate the reflections from polycrystalline ZnS phase. No epitaxial relationship between the ZnS and the ZnO was found. Epitaxial relationship between ZnO and ZnS was frequently found in some high temperature synthesized ZnO@ZnS samples18–21 in spite of a large mismatch between ZnO and ZnS structures. In this work, ZnS NPs are uniformly and randomly distributed on ZnO core surface because of the low atomic mobility at low temperature. Fig. 6 shows EDX mapping of the ZnO@ZnS CSNPs prepared with 0.05 M concentration of Na2S. It is clear that increasing the concentration of the S2− ions leads to formation of the more uniform and dense shell. Also, no ZnS NPs was observed in this concentration.
 |
| | Fig. 4 (a) Bright field TEM image of a ZnO@ZnS CSNP, (b) corresponding SAED pattern from (a). (c) EDX mapping exhibiting S, O and Zn distribution, (d) HRTEM image from the rectangle region on the left of (a), and (e) FFT pattern from (d). | |
 |
| | Fig. 5 (a) Bright field TEM image of pure ZnO NPs. (b) STEM image of ZnO@ZnS CSNP. | |
 |
| | Fig. 6 EDX mapping of the ZnO@ZnS CSNPs prepared with 0.1 M concentration of Na2S solution. | |
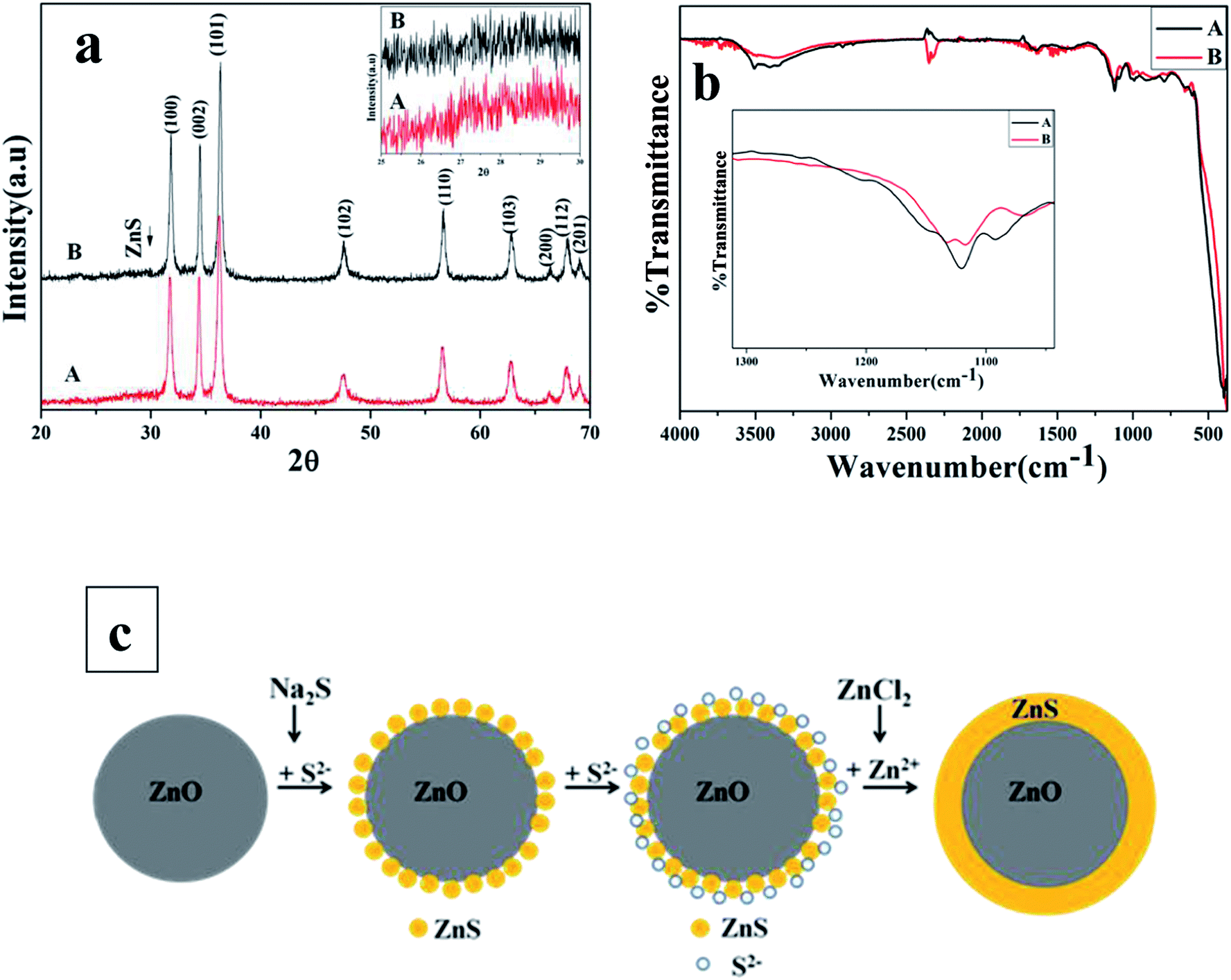
In order to investigate the ZnO@ZnS CSNPs formation mechanism, synthesis of the ZnO@ZnS CSNPs prepared with 0.05 M concentration of Na2S was repeated without addition of the ZnCl2. The result was compared with the case when ZnCl2 was added. Fig. 7a shows the XRD spectra of the ZnO@ZnS CSNPs with (denoted by A) and without (denoted by B) the addition of ZnCl2, respectively. It can be seen in Fig. 7a that the intensity of some peaks belonging to the ZnO are higher for the case without addition of the ZnCl2, which can be due to the covering of the ZnO NPs with a thicker shell. Also, a magnified XRD spectrum from 25° to 30° is shown as insert and indicating that the peak intensity belonging to the ZnS is higher for the sample prepared by the addition of the ZnCl2. Further information was obtained by studying the FTIR spectra for both cases. Fig. 7b shows the FTIR spectra of the ZnO@ZnS CSNPs with (denoted by A) and without (denoted by B) the addition of ZnCl2, respectively. It is clear that the characteristic ZnS vibration peak is higher in the ZnO@ZnS CSNPs prepared by the addition of the ZnCl2. This means that the number of Zn–S bands in the ZnO@ZnS CSNPs grown by adding the ZnCl2 has increased. Based on this we propose that the growth mechanism of fully covered ZnO@ZnS CSNPs in our procedure is as follows:
 |
| | Fig. 7 (a) XRD spectra of ZnO@ZnS CSNPs prepared (a) without and (b) with adding ZnCl2. (b) ATR-FTIR spectra, and (c) A schematic illustration of the ZnO@ZnS CSNPs formation mechanism. | |
After dispersing the ZnO NPs in isopropanol, the pH of the solution was adjusted to around 10. As the ZnO NPs are soluble in acidic and basic media, the surface of the NPs will start dissolve and the S2− released from the Na2S which is added drop wise, will react with the Zn2+ and leads to produce ZnS around the ZnO NPs:
The formation of a layer of ZnS separates the ZnO NPs from the sulfur ions in the solution so that no more reaction could proceed between the two. By adding the ZnCl2 to the mixture drop wise, the unreacted S2− ions around the shell will react with the Zn2+ from the solution forming more ZnS on the surface of the NPs. The formation mechanism of ZnO@ZnS CSNPs is shown in the schematic diagram of Fig. 7c.
In the work reported by Sharma et al.,25 before adding the nutrients to form the ZnO NPs the two ZnCl2 and Na2S solutions were mixed together, which leads to formation of ZnS nanoparticles in the solution before adding the ZnO NPs. Moreover, they did not explain how the ZnO NPs are covered with ZnS. To prevent the formation of any impurity in our work, Na2S solution was added to ZnO NPs drop wise and it was kept under continuous stirring for 2 h. After some of the S2− ions have reacted with the surface of ZnO NPs in the solution, the ZnCl2 solution is added to react with the unreacted S2− around the shell and form a dense shell with full coverage. Moreover, in our work all mixed solutions were added drop wise, and all mixtures were under stirring all the time so that the ions could be dispersed uniformly in the mixture and form identical particles. Hence, our sequence of the nutrients introduction to the synthesis of the ZnO@ZnS CSNPs has been rational and has resulted in full coverage.
3.2 Photocatalytic performance
Degradation of the dye pollutant in photocatalytic decomposition process takes place via photo-generation of electron–hole pair between conduction and valence bands. The photo-generated holes (h+) in the valence band react with either H2O or OH− to produce the HO˙ through the following reactions:34while the electrons (e−) in the conduction band react with the adsorbed O2 on the ZnO surface to generate O2˙− and according to the following steps, it lead to the generation of HO˙ radicals:| | | e− + O2 → O2˙− + H+ → HO2˙ + O2˙− → HO2˙ + O− | (3) |
| | | H2O2 + O2˙− → HO˙ + OH− + O2 | (5) |
Finally, the dye pollutant is decomposed by the generated agents:34
| | | Dye + (O2˙− or HO˙ or OHO2˙) → intermediate → product | (8) |
Since the point of zero charge of a photocatalyst varies with pH, the adsorption of dye on the surface of the photocatalyst is different at various pH values. It is important to mention that the decomposition of some dyes occurs on the surface of the photocatalyst, so the adsorption of the dye is a crucial step in a photocatalytic degradation process. Although the dye with high adsorption degrades faster, the effect sites for absorbing the UV light decreases with increasing the adsorption of the dye on the surface of the photocatalyst. Moreover, higher pH value could provide higher concentration of hydroxyl ions to react with holes and form hydroxyl radicals which subsequently cause an increased photocatalytic degradation rate. The combined effect of the adsorption onto the photocatalyst and concentration of the hydroxyl radicals will determine the extent of the degradation.35
The chemical structure of the Rose Bengal dye is shown in Fig. 8. The Rose Bengal becomes negatively charged by losing the Na+ after dissolving in water and it can thus be adsorbed on positively charged surfaces. Initially, the dye adsorption on the surface of the ZnO NPs and the ZnO@ZnS CSNPs (ZnO@ZnS1 (0.025 M) ZnO@ZnS2 (0.05 M) and ZnO@ZnS3 (0.1 M)) was investigated at three different pH values (4, 7, and 10) and the results are summarized in Fig. 9 and according to this column chart, the maximum and minimum adsorption of the Rose Bengal takes place at pH of 4 and 10, for the ZnO@ZnS CSNPs and ZnO NPs, respectively and there was no adsorption on the surface of the ZnO NPs at pH of 10. Another important point which can be found from Fig. 8 is that the dye is adsorbed more on the surface of the ZnO@ZnS1 CSNPs and ZnO@ZnS2 CSNPs at each of the three pH values. This is because of the more defects and dangling bonds on the surface, which can act as traps for these two samples. To confirm this claim the photoluminescence spectra of each samples was prepared and is shown in Fig. 10. Table 1 shows the ratio of the near band edge (NBE) to the deep level emission (DLE) intensity of the all samples and indicate that the defects are decreasing gradually by filling up the oxygen vacancies via S atoms for the case of ZnO@ZnS1, and then it is increased even further for the ZnO@ZnS2 and for ZnO@ZnS3 is decreased again. Coating the surface of the particles with ZnS shell leads to filling up of the oxygen vacancies on the surface of the ZnO NPs with the S atoms. But for the ZnO@ZnS1 the concentration of the sulfur source is not enough to form a full shell, therefore the S atoms fill up some oxygen vacancies and decrease the DLE intensity. Although a lot of oxygen vacancies have been filled in ZnO@ZnS2 sample, its ZnS layer has more sulfur vacancies. With further increasing of the sulfur ions, a more uniform ZnS layer sediments on the ZnO NPs and forms a shell with fewer defects.
 |
| | Fig. 8 Chemical structure of Rose Bengal dye. | |
 |
| | Fig. 9 Adsorption of Rose Bengal on the surface of ZnO NPs and ZnO@ZnS CSNPs. | |
 |
| | Fig. 10 PL spectra of ZnO NPs and ZnO@ZnS CSNPs. | |
Table 1 The NBE to DLE emission intensity of the samples
| Sample |
NBE to DLE ratio |
| ZnO |
0.18 |
| ZnO@ZnS1 |
0.25 |
| ZnO@ZnS2 |
0.08 |
| ZnO@ZnS3 |
0.6 |
Fig. 11–13 show the photocatalytic removal of the Rose Bengal using ZnO NPs and ZnO@ZnS CSNPs after 120 min. and at three different pH values: 4, 7, and 10, respectively. More detailed results are shown in the plot of C/C0versus time, which is given in Fig. 14, where C0 is the initial concentration of the dye, C is the concentration of the dye after irradiation time t in min. It can be seen from Fig. 12a that, at a pH value of 4, almost 35%, 33%, 51%, and 37% of dye was degraded after 120 min illumination for ZnO NPs, ZnO@ZnS1, ZnO@ZnS2, and ZnO@ZnS3, respectively. According to these results, the photocatalytic activity of ZnO@ZnS2 and ZnO@ZnS3 is better than the ZnO NPs and the ZnO@ZnS2 with the most adsorption of dye performed better as a photocatalyst at pH 4. The plot of C/C0versus time for a pH of 7, namely Fig. 14b, indicates that the dyes was decomposed by 49%, 21%, 30%, and 28% for the ZnO NPs, ZnO@ZnS1, ZnO@ZnS2, and ZnO@ZnS3, respectively, and indicate that the ZnO NPs show better photocatalytic activity at this pH compared to the CSNPs. According to Fig. 14c, the Rose Bengal dye was degraded by about 50%, 35%, 50%, and 38% for ZnO NPs, ZnO@ZnS1, ZnO@ZnS2, and ZnO@ZnS3, respectively and shows almost the same photocatalytic activity for ZnO NPs and ZnO@ZnS1. Using graphene/polyaniline composite, Rose Bengal degradation was reported.36 In this work they employed a xenon arc lamp 300 W for 3 h. and they reported 50% dye removal. In our case we have used a 62 W UV lamp and achieved a 50% Rose Bengal degradation in 2 h. This shows that the presented use of ZnO@ZnS CSNPs as Rose Bengal degradation is efficient. The kinetics of the photocatalytic degradation of organic dyes usually follows the Langmuir–Hinshelwood mechanism:37
| | | r = −dC/dt = (kKC)/(1 + KC), | (9) |
where
r is the degradation rate of the reactant (mg L
−1 min
−1),
C is the concentration of the reactant (mg L
−1),
k is the reaction rate constant (mg L
−1 min
−1),
K is the adsorption constant of the reactant (L mg
−1). When
C is very small, the equation can be simplified to:
| | | ln(C0/C) = kKt = Kappt, | (10) |
where
Kapp is the apparent first-order rate constant given by the slope of the graph of ln(
C0/
C)
vs. t and
C0 is the initial concentration of the reactant. The plots of ln(
C0/
C)
versus time for ZnO NPs and ZnO@ZnS CSNPs at the three different pH values is shown in
Fig. 15 which is in agreement with the Langmuir–Hinshelwood first-order kinetic behavior.
 |
| | Fig. 11 UV-vis absorption spectra of Rose Bengal dye solution in 30 min intervals at pH 4. | |
 |
| | Fig. 12 UV-vis absorption spectra of Rose Bengal dye solution in 30 min intervals at pH 7. | |
 |
| | Fig. 13 UV-vis absorption spectra of Rose Bengal dye solution in 30 min intervals at pH 10. | |
 |
| | Fig. 14 The plot of C/C0versus reaction time at (a) pH 4, (b) pH 7, and (c) pH 10. | |
 |
| | Fig. 15 The plot of ln(C0/C) versus time at (a) pH 4, (b) pH 7, and (c) pH 10. | |
To explain the possible causes of the observed results, we note that, covering the ZnO NPs with ZnS leads to form a type II system. In type II systems, the effective band gap of the core–shell system is smaller than both the core and the shell material as it is clear in Fig. 16. Smaller band gap needs lower energy to produce electron–hole pairs and also leads to a more effective transfer of electrons between the core and the shell in addition to a relatively long lifetime of the photogenerated electron–hole pairs.38 These conditions lead us to the finding that the ZnO@ZnS CSNPs can show better photocatalytic efficiency, but in some cases electron–hole recombination occurs at the interface between the core and the shell material and this can reduce the photocatalytic activity of core–shell nanostructures.
 |
| | Fig. 16 Schematic illustration of dye degradation over ZnO@ZnS CSNPs. | |
The effect of the pH on the efficiency of the dye decolorization is difficult to assess because of the multiple roles of the pH of the solution. First, there is a possibility of the deionization state of the surface which influence the adsorption of dye onto the nanocatalyst. Second, the hydroxyl radicals, which are essential for photocatalytic process, can be formed by reaction between the positive holes and the hydroxyl ions. Positive holes are major species at low pH and hydroxyl ions are major at natural and high pH values. Thirdly, at low pH values the ZnO nanoparticles tend to agglomerate so that its surface will be decrease and this result in reduction of photocatalytic activity.39
All the above mentioned factors play a role in determining the photocatalytic efficiency and by considering all of them the photocatalytic behavior of one nanocatalyst at a specific pH is justified. According to the obtained results in this paper the ZnO@ZnS CSNPs show better photocatalytic efficiency at a pH of 4 compared to ZnO NPs. The possible reason behind this could be explained by the fact that the ZnO NPs agglomerate at this pH and this results in a reduction of effective surface for absorbing the UV light and also the adsorption of the dye while in ZnO@ZnS CSNPs, the holes which are the dominant species at low pH are even more due to the transferred holes from the core to the shell leading to an increase of the hydroxyl radicals. Also, as it was mentioned before the adsorption of the dye onto the surface of the ZnO@ZnS CSNPs is maximum at a pH of 4. This can also improve the photocatalytic activity. At a pH of 7 ZnO NPs show better photocatalytic behavior compared to the ZnO@ZnS CSNPs. ZnO NPs are not agglomerated at pH 7 and it seems that this increases the effective sights for absorbing UV light, which leads to improvement of its photocatalytic behavior compared to a pH of 4. For the ZnO@ZnS CSNPs, the photocatalytic activity has declined compared to pH 4, which is due to decreasing the adsorption of the dye onto the surface of the ZnO@ZnS CSNPs. At a pH 10 the photocatalytic behavior of the two photocatalysts is almost the same. It seems that at a pH of 10 the effective factor is increasing the amount of hydroxyl radicals. Increasing the amount of hydroxyl ions by increasing the pH, the higher concentration of the hydroxyl ions leads to higher concentration of hydroxyl radicals, which causes an enhanced photocatalytic decolorization rate.
4. Conclusions
ZnO NPs were prepared with an efficient co-precipitation method without any surfactant and then covered with ZnS in two steps using a chemical method at low temperature. XRD and FTIR spectra show the formation of ZnS and the Na2S solution concentration on the reaction result. SEM analysis of ZnO@ZnS CSNPs shows that increasing the sulfur concentration leads to the agglomeration of particles and also confirms that the morphology of ZnO nanoparticles is not entirely spherical also HRTEM shows that the grain surface is quite rough. EDX mapping demonstrates a full coverage of ZnO@ZnS core–shell nanostructures. HRTEM also shows that the ZnS shell consists of nano-crystals of ZnS and no epitaxial relationship was found between ZnO and ZnS because of low growth temperature. The photocatalytic activity of ZnO@ZnS CSNPs was examined to degrade Rose Bengal dye at different pH values and compared to that of ZnO NPs. Although ZnO NPs perform as a more active photocatalyst at pH 7, the ZnO@ZnS CSNPs are preferable for degradation at a pH of 4. Our results of 50% degradation of Rose Bengal dye with only 62 W and 2 h of illumination is superior to using existing graphene/polyaniline composites. Thus, our CSNPs can be used as an efficient degradation agent.
Acknowledgements
Azar Sadollahkhani would like to thank Shahid Chamran University and Linkoping University for financial support.
References
- I. Kazeminezhad and A. Sadollahkhani, Mater. Lett., 2014, 120, 267 CrossRef CAS PubMed.
- M. Chen, L. Hu, J. Xu, M. Liao, L. Wu and X. Fang, Small, 2011, 7, 2449 CAS.
- Y. Kim and S. Kang, Acta Mater., 2011, 59, 3024 CrossRef CAS PubMed.
- J. Fan, A. Shavel, R. Zamani, C. Fabrega, J. Rousset, S. Haller, F. Guell, A. Carrete, T. Andreu, J. Arbiol, J. Ramon Morante and A. Cabot, Acta Mater., 2011, 59, 6790 CrossRef CAS PubMed.
- K. Vignesh, A. Suganthi, M. Rajarajan and S. A. Sara, Powder Technol., 2012, 224, 331 CrossRef CAS PubMed.
- B. Xiang, P. Wang, X. Zhang, S. A. Dayeh, D. P. R. Aplin, C. Soci, D. Yu and D. Wang, Nano Lett., 2007, 7, 323 CrossRef CAS PubMed.
- P. Liu, Y. Li, Y. Guo and Z. Zhang, Nanoscale Res. Lett., 2012, 7, 220 CrossRef PubMed.
- W. Lu, C. Jiang, D. Caudle, C. Tang, Q. Sun, J. Xu and J. Song, Phys. Chem. Chem. Phys., 2013, 15, 13532 RSC.
- X. Xu, M. Wu, M. Asoro, P. J. Ferreira and D. L. Fan, Cryst. Growth Des., 2012, 12, 4829 CAS.
- M. G. Nair, M. Nirmala, K. Rekha and A. Anukaliani, Mater. Lett., 2011, 65, 1797 CrossRef CAS PubMed.
- P. Reiss, M. Protiere and L. Li, Small, 2009, 5, 154 CrossRef CAS PubMed.
- Z. Zeng, C. S. Garoufalis, A. F. Terzis and S. Baskoutas, J. Appl. Phys., 2013, 114, 23510 CrossRef PubMed.
- J. Zhaia, X. Taoa, Y. Pua, X. F. Zenga and J. F. Chenb, Appl. Surf. Sci., 2010, 257, 393 CrossRef PubMed.
- A. Roychowdhury, S. Prakash Pati, S. Kumar and D. Das, Powder Technol., 2014, 254, 583 CrossRef CAS PubMed.
- S. Sahare, S. J. Dhoble, P. Singh and M. Ramrakhiani, Adv. Mater. Lett., 2013, 4, 169 Search PubMed.
- X. Fang, Y. Bando, M. Liao, U. K. Gautam, C. Zhi, B. Dierre, B. Liu, T. Zhai, T. Sekiguchi, Y. Koide and D. Golberg, Adv. Mater., 2009, 21, 2034 CrossRef CAS PubMed.
- W. H. Nam, Y. S. Lim, W. S. Seo and J. Y. Lee, Philos. Mag., 2013, 93, 4221 CrossRef CAS.
- X. Zhang, M. L. Chen, J. Wen, L. L. Wu, H. Gao and D. Zhang, CrystEngComm, 2013, 15, 1908 RSC.
- J. D. Fan, C. Fábrega, R. Zamani, A. Shavel, F. Güell, A. Carrete, T. Andreu, A. M. López, J. R. Morante, J. Arbiol and A. Cabot, J. Alloys Compd., 2013, 555, 213 CrossRef CAS PubMed.
- B. H. Ahn and J. Y. Lee, CrystEngComm, 2013, 15, 6709 RSC.
- D. D. Lin, H. Wu, R. Zhang, W. Zhang and W. Pan, J. Am. Ceram. Soc., 2010, 93, 3384 CrossRef CAS PubMed.
- Y. H. Mun, S. H. Park, H. S. Ko and C. M. Lee, J. Korean Phys. Soc., 2013, 63, 1595 CAS.
- S. K. Panda, A. Dev and S. Chaudhuri, J. Phys. Chem. C, 2007, 111, 5039 CAS.
- W. H. Nam, Y. S. Lim, W. S. Seo, H. K. Cho and J. K. Lee, J. Nanopart. Res., 2011, 13, 5825 CrossRef CAS.
- S. Sharma and S. Chawla, Electron. Mater. Lett., 2013, 9, 267 CrossRef CAS.
- S. Liu, X. Wang, W. Zhao, K. Wang, H. X. Sang and Z. He, J. Alloys Compd., 2013, 568, 84 CrossRef CAS PubMed.
- S. P. Buthelezi, A. O. Olaniran and B. Pillay, Molecules, 2012, 17, 14260–14274 CrossRef CAS PubMed.
- Y. Liao, C. Xie, Y. Liu, H. Chen, H. Li and J. Wu, Ceram. Int., 2012, 38, 4437–4444 CrossRef CAS PubMed.
- S. Suwanboon, P. Amornpitoksuk, A. Sukolrat and N. Muensit, Ceram. Int., 2013, 39, 2811–2819 CrossRef CAS PubMed.
- T. Madrakian, A. Afkhami and M. Ahmadi, Spectrochim. Acta, Part A, 2012, 99, 102–109 CrossRef CAS PubMed.
- I. A. Siddiquey, T. Furusawa, M. Sato, N. M. Bahadur, M. M. Alam and N. Suzuki, Ultrason. Sonochem., 2012, 19, 750–755 CrossRef CAS PubMed.
- G. Manjula Nair, M. Nirmala, K. Rekha and A. Anukaliani, Mater. Lett., 2011, 65, 1797–1800 CrossRef PubMed.
- S. M. Lam, J. C. Sin, A. Z. Abdullah and A. R. Mohamed, Desalin. Water Treat., 2012, 41, 131–169 CrossRef CAS.
- U. G. Akpan and B. H. Hameed, J. Hazard. Mater., 2009, 170, 520–529 CrossRef CAS PubMed.
- L. Gomathi and K. Mohan Reddy, Appl. Surf. Sci., 2010, 256, 3116–3121 CrossRef PubMed.
- S. Ameen, H. K. Seo, S. M. Akhtar and H. S. Shin, Chem. Eng. J., 2012, 210, 220–228 CrossRef CAS PubMed.
- S. M. Lam, J. C. Sin, A. Z. Abdullah and A. R. Mohamed, Desalination, 2012, 41, 131–169 CAS.
- M. Y. Lu, J. Song, M. P. Lu, C. Y. Lee, L. J. Chen and Z. L. Wang, ACS Nano, 2009, 3, 357–362 CrossRef CAS PubMed.
- W. Chen, H. Ruan, Y. Hu, D. Li, Z. Chen, J. Xian, J. Chen, X. Fu, Y. Shao and Y. Zheng, CrystEngComm, 2012, 14, 6295–6305 RSC.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a