
Electronic and optical properties of chair-like and boat-like graphane
Abstract
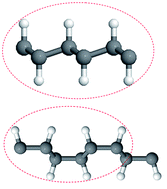
We have studied two favorable conformations of graphane, the chair-like graphane and boat-like graphane. In the chair-like, the H atoms attached to the C atoms alternate on both sides of the sheet while in the boat-like, the C bonded H atoms alternate in pair. Both conformations of graphane have a 2D puckered honeycomb like structure with one hydrogen atom bonded covalently (sp3) to each carbon atom. The chair-like belongs to the P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m1 (164) space group and the boat-like belongs to the Pmmn (59) space group. We have used the state-of-the-art full potential linear augmented plane wave (FPLAPW) method with different possible approximations for the exchange–correlation (XC) potential. The XC potential was described by the local density approximation (LDA) of Ceperley–Alder (CA), the generalized gradient approximation (GGA) of Perdew–Becke–Ernzerhof (PBE) and the Engel–Vosko generalized gradient approximation (EVGGA). The calculated partial density of states for both configurations, show that there exists a strong hybridization between C and H orbitals which confirms the existence of the covalent bonds. The electronic charge density distribution of both configurations (chair-like and boat-like) has been calculated, the charge accumulates along C–C and C–H bonds. According to the electro-negativity values of C (2.55) and H (2.1), it is clear that there is strong covalent bonding between C and H atoms. The linear optical properties give a deep insight into the electronic structure. The calculated values of the energy gap and the bond lengths show good agreement with previous results.
m1 (164) space group and the boat-like belongs to the Pmmn (59) space group. We have used the state-of-the-art full potential linear augmented plane wave (FPLAPW) method with different possible approximations for the exchange–correlation (XC) potential. The XC potential was described by the local density approximation (LDA) of Ceperley–Alder (CA), the generalized gradient approximation (GGA) of Perdew–Becke–Ernzerhof (PBE) and the Engel–Vosko generalized gradient approximation (EVGGA). The calculated partial density of states for both configurations, show that there exists a strong hybridization between C and H orbitals which confirms the existence of the covalent bonds. The electronic charge density distribution of both configurations (chair-like and boat-like) has been calculated, the charge accumulates along C–C and C–H bonds. According to the electro-negativity values of C (2.55) and H (2.1), it is clear that there is strong covalent bonding between C and H atoms. The linear optical properties give a deep insight into the electronic structure. The calculated values of the energy gap and the bond lengths show good agreement with previous results.
 Please wait while we load your content...
Please wait while we load your content...

