
Theoretical study of CO catalytic oxidation on free and defective graphene-supported Au–Pd bimetallic clusters
Wei Zhanga,
Daojian Cheng*a and
Jiqin Zhu*b
aState Key Laboratory of Organic–Inorganic Composites, Beijing University of Chemical Technology, Beijing 100029, China. E-mail: chengdj@mail.buct.edu.cn
bState Key Laboratory of Chemical Resource Engineering, Beijing University of Chemical Technology, 100029 Beijing, China. E-mail: zhujq@mail.buct.edu.cn
First published on 26th August 2014
Abstract
In this work, CO adsorption and oxidation on free and defective graphene-supported AumPdn (m + n = 13) clusters with either icosahedral (ICO) or truncated octahedral (TO) structures are investigated using density functional theory calculations. It is found that CO adsorbs more strongly than molecular O2 on the surface of these clusters, which is attributed to the strong hybridization between metal-d and C-p orbitals. In addition, the structural transformation from ICO to TO is observed for the Au-rich clusters upon CO and O2 adsorption because the stability of the ICO cluster is lower than that of the TO one. It is also found that the free Au12Pd1 cluster with TO structure exhibits the lowest energy barrier for CO oxidation among the free clusters (0.17 eV). When the Au12Pd1 cluster is supported on the single vacancy defective graphene, an increase in stability, a decrease in the adsorption strength of CO and O2, and a slight increase in the energy barrier (0.41 eV) for CO oxidation compared to the corresponding free cluster are observed. Our results are expected to be useful for future applications of graphene-supported bimetallic nanocatalysts.
I. Introduction
The fascinating catalytic properties of metal clusters stem from their non-crystalline configurations, large surface-to-volume ratio, and finite size effect.1 One of the most famous examples is gold; although gold has long been regarded as an inert metal, the pioneering work of G. J. Hutchings and Haruta et al. demonstrated that gold nanoparticles are exceptionally active catalysts towards a number of reactions.2–5 The activity of Au clusters for CO oxidation is strongly dependent on the cluster size,6 cluster shape,7 surface coating,8 and oxide support.9 In recent years, bimetallic clusters have attracted considerable attention since the introduction of a second metal into the single metal cluster can improve the catalytic activities of the clusters by the synergetic effect.10 Notably, Au-based bimetallic clusters prepared by the mixing of Au with other metals (Pd, Pt, or Cu)11–14 have been used to improve the catalytic activities, stabilities, and selectivities of pure Au clusters for CO oxidation.15–18 For example, Scott and co-workers found that the supported Au–Pd clusters exhibit higher catalytic activities towards CO oxidation compared with the supported Au-only or Pd-only catalysts.19Graphene, a one-atom-thick carbon sheet with unique electronic and geometric properties, is regarded as one of the most promising candidates for the next generation of electronic materials.20–22 Recently, graphene has been extensively studied as a support for heterogeneous catalysts due to its huge surface to-volume ratio for the catalytic reaction.23–27 The presence of carbon vacancies on graphene can be used as anchoring points for the binding of metal clusters. For example, the binding of Pt, Fe, Pt, and Al metal clusters with graphene28–30 can be enhanced by the dangling sp2 carbon bonds at the monovacancy sites of defective graphene, significantly enhancing the stabilities of metal clusters.31 In addition, the defects in graphene can promote the adsorption of small molecules on metal clusters.28 Therefore, the defective graphene-supported Au–Pd bimetallic clusters are anticipated to be very active catalysts for CO oxidation, and further studies are needed to verify this expectation.
Density functional theory (DFT)32 calculations have been used to evaluate the catalytic properties of metal clusters for CO oxidation. In a previous work, DFT calculations were performed to study CO oxidation on Au–Pd bimetallic surfaces. Zhang et al.33 used DFT to study the catalytic properties of CO oxidation on Au/Pd (111) surfaces and found that the system with Au on the surface of a Pd16Au4 slab exhibited the lowest energy barrier for CO catalytic oxidation. On the other hand, Yuan et al.34 investigated CO oxidation on Pd-decorated Au (111) surfaces, finding that the Pd2-decorated Au (111) surface was highly reactive for CO oxidation. In addition, free Au/Pd core–shell clusters have been found to be highly efficient catalysts for CO oxidation by DFT calculations.35,36 However, to the best of our knowledge, a theoretical understanding of the reaction mechanism of CO oxidation on graphene-supported Au–Pd bimetallic clusters is still lacking,37 and the details of how the support and cluster shape affect the reaction mechanism are also unknown.
In this paper, we apply DFT calculations to investigate CO adsorption and oxidation on free and defective graphene-supported AumPdn (m + n = 13) clusters with either icosahedral (ICO) or truncated octahedral (TO) structures. For the free AumPdn (m + n = 13) clusters, we study the relatively stable structures of 13-atom Au–Pd clusters as well as O2 and CO adsorption, the co-adsorption of O2 and CO, and CO oxidation on these clusters. We compare the energy barrier for CO oxidation on the defective graphene-supported Au12Pd1 cluster with that of CO oxidation on the corresponding free cluster.
II. Computational details
In this work, 13-atom Au–Pd bimetallic clusters with icosahedral (ICO) and truncated octahedral (TO) structures are studied. From the structural point of view, attention has usually been concentrated on high-symmetry configurations,38 which are often found to be the lowest-energy structures of Au–Pd bimetallic clusters at the DFT level. The ICO or TO structures are good candidates for smaller clusters due to their more favourable surface energies at the expense of introducing internal strain.39 The lowest-energy atomic ordering of such clusters is calculated at the empirical potential level40–42 and then subjected to local relaxation by DFT. In the calculations, an EAM empirical potential43–45 is used to model the metal–metal interactions; its explicit form43,44,46 and parameters for the Au–Pd systems46,47 can be found elsewhere. The resulting configurations are used to study the adsorption and reaction properties. Fig. 1 shows the equilibrium geometries of the four free metal clusters with ICO and TO structures used in this work.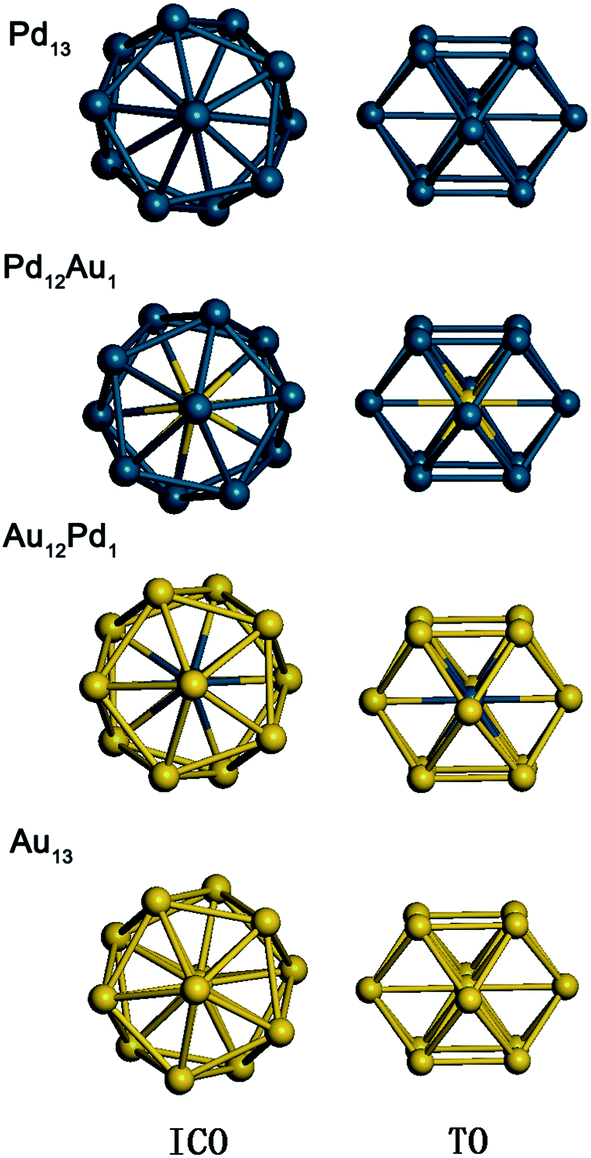 | ||
| Fig. 1 The configurations of Pd13, Pd12Au1, Au12Pd1, and Au13 with icosahedral (ICO) and truncated octahedral (TO) structures. Au and Pd atoms are represented by yellow and blue spheres, respectively. | ||
DFT calculations were performed using the PWSCF (plane-wave self-consistent field) plane wave code in the Quantum ESPRESSO package.48 All the calculations were carried out by the spin-polarized generalized gradient approximation (GGA) with the Perdew–Burke–Ernzerhof (PBE)49 xc-functionals and ultrasoft pseudopotentials.50 It is noted that ultrasoft pseudopotentials have been used successfully to investigate the chemical properties of many transition metal systems including Au–Pd systems.50–53 Spin-polarized calculations were performed using values of 40 and 400 Ry as the energy cut-offs for the selection of the plane waves describing the wave function and the electronic density, respectively. For free metal clusters, a rectangular supercell with a size of 30 × 30 × 30 Å3 was employed in the calculations. The first Brillouin zone was sampled at the Gamma-point, and the electronic levels were broadened using a Gaussian smearing technique with a smearing parameter of 0.002 Ry. The geometry of the cluster upon or without adsorption is optimized until the total energy converges to 10−6 eV. For the Au12Pd1 cluster supported on defective graphene, an orthorhombic supercell of 14.76 × 14.76 × 31.51 Å3 with periodic boundary conditions was used. After checking for the possibility of one, two, three and four atom conformations with defective graphene, only the configuration with one metal atom interacting with the graphene cluster was found to be stable. The Au12Pd1–graphene system was separated from its periodic images in the z direction by a vacuum space of 25 Å. The integration of the Brillouin zone was conducted using a 2 × 2 × 1 Monkhorst–Pack grid with the K-points for the Au12Pd1–graphene system. In this work, the van der Waals (vdW) interaction54 is not included since the vdW interaction has only a slight effect on the adsorption and energy barrier energies for CO oxidation on metal systems.24,26,27
We calculated the adsorption energies according to the equation:
| ΔEads = Etotal − Ecluster − Eadsorbate | (1) |
Furthermore, the co-adsorption energies of CO with molecular or atomic oxygen were calculated as:
| ΔEcoads = Etotal − Ecluster(or cluster+graphene) − ECO − EO2(or O) | (2) |
In the above equations, Etotal, Ecluster (or cluster+graphene), Eadsorbate, ECO and EO2 (or O) correspond to the electronic energies of the adsorbed species on the cluster, the bare cluster (or the bare cluster on graphene), a gas-phase adsorbate, gaseous CO and gaseous O2 (or O), respectively. The climbing-image nudged elastic band (NEB) method55 was adopted here to determine the reaction pathway for CO oxidation on Au–Pd clusters. The reaction paths are interpolated by fitting a cubic polynomial through all images on each path guided by the force tangent to the reaction coordinate at each image. Once a minimum-energy path is determined, the transition state is located.56
III. Results and discussion
A. CO and O2 adsorption on free clusters
The calculated average bond lengths, average binding energies, and d-band centres of Pd13, Pd12Au1, Au12Pd1, and Au13 with ICO and TO structures are shown in Table 1. The average bond length increases with increasing Au concentration for clusters with the same morphology because Au is larger than Pd. In addition, the average bond lengths of the TO structures are smaller than those of the ICO structures for free clusters with the same composition. The average binding energy corresponds to the stability of the cluster. As listed in Table 1, the absolute value of the average bonding energy for the Pd-rich clusters follows the order TO < ICO, while the absolute value of the average bonding energy of TO is bigger than that of ICO for the Au-rich clusters. Thus, the ICO structure is more stable than the TO structure for Pd-rich clusters, while the opposite order is found for Au-rich clusters. Relatively stable structures should be used to study the catalytic properties of the clusters due to the structural interconversion between isomers in the working environment. For example, the ICO to TO structural transformation in Au-rich clusters upon O2 adsorption is observed because the stability of the ICO cluster is lower than that of the TO structure for Au-rich clusters (Fig. 2). Thus, the following paragraphs only discuss the adsorption, co-adsorption and CO oxidation reaction on ICO Pd12Au1 and Pd13 clusters and TO Au12Pd1 and Au13 clusters.| Cluster | Pd13 | Pd12Au1 (Au in the core) | Au12Pd1 (Pd in the core) | Au13 | ||||
|---|---|---|---|---|---|---|---|---|
| Isomer | ICO | TO | ICO | TO | ICO | TO | ICO | TO |
| Length | 3.22 | 2.72 | 3.25 | 2.73 | 3.34 | 2.82 | 3.37 | 2.84 |
| Eb (eV per atom) | −2.29 | −2.24 | −2.23 | −2.22 | −1.99 | −2.07 | −1.89 | −1.96 |
| Ed (eV) | −2.28 | — | −1.51 | — | — | −2.73 | — | −2.74 |
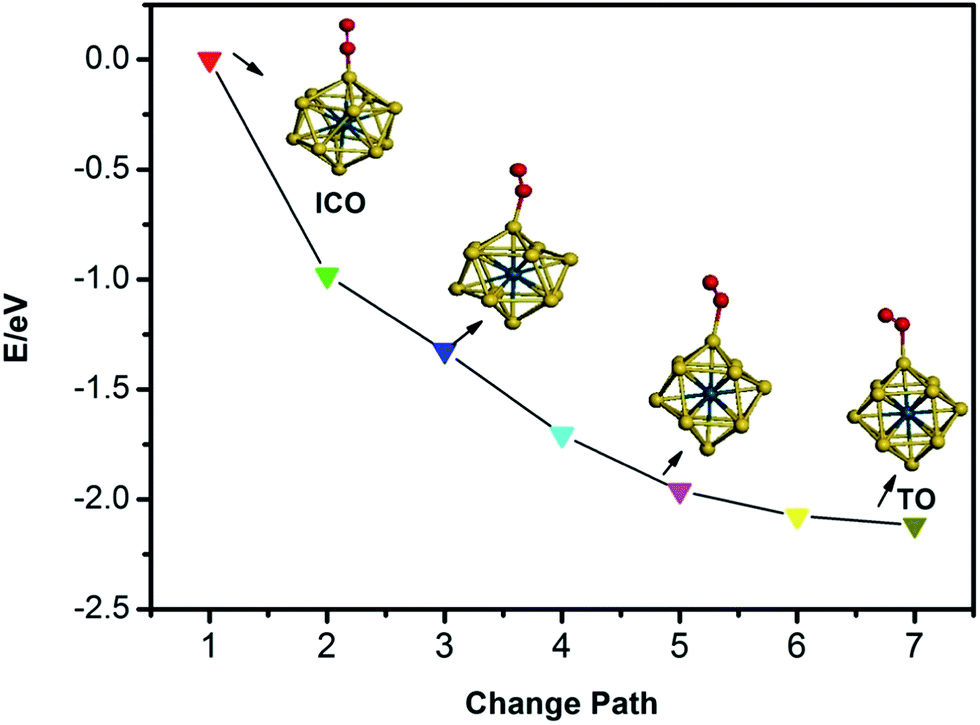 | ||
| Fig. 2 The structural transformation from icosahedron to truncated octahedron for the free Au12Pd1 cluster upon O2 adsorption. | ||
In the ICO structure, there are 20 equivalent triangular fcc (111)-like facets (Fig. 3). In the TO structure, the facets consist of both triangles and quadrangles (see Fig. 3). It is found that CO and O2 cannot be adsorbed on the (100) hollow site in the quadrangle of the TO structure. To compare the Pd-rich clusters with the ICO structure, only the triangular facet was chosen to study CO adsorption and oxidation on Au-rich clusters. For CO adsorption, the molecule prefers the “end-on” configuration with the C atom binding to the Pd atoms (see Fig. 3).57 Three possible sites were calculated: on the top (T), on the bridge (B), and on the hollow (H). We have calculated the energies of CO adsorption at these sites on the Pd13, Pd12Au1 (Au in the centre), Au12Pd1 (Pd in the centre), and Au13 clusters. The results are summarized in Table 2. H is the most stable adsorption site for Pd13 and Pd12Au1 clusters, with adsorption energies of −2.56 and −2.44 eV, respectively, while T (vertex) is the most stable site for Au12Pd1 and Au13 clusters, with adsorption energies of −1.41 and −1.36 eV, respectively.
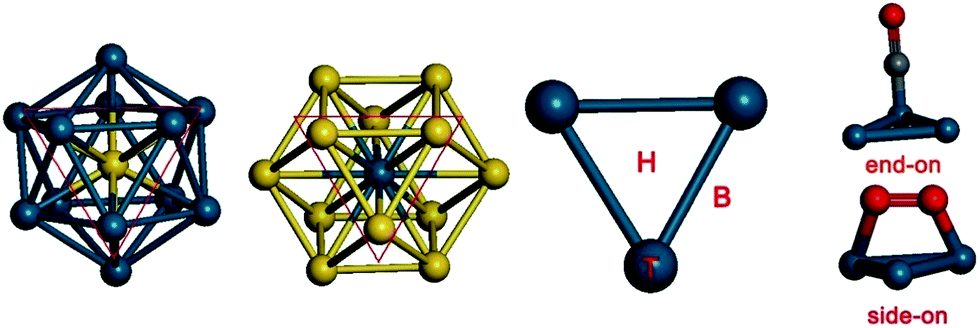 | ||
| Fig. 3 The possible adsorption sites for CO and O2 molecules on the icosahedral (ICO) and truncated octahedral (TO) clusters. | ||
| Cluster | Adsorption site | |||||
|---|---|---|---|---|---|---|
| T | B | H | B1 | T-H | ||
| Pd12Au1 | O2 | −0.71 | −0.73 | −0.64 | −1.44 | −1.58 |
| CO | −1.84 | −2.33 | −2.56 | U | U | |
| Pd13 | O2 | −0.78 | −0.57 | −0.55 | −1.34 | −1.43 |
| CO | −1.35 | −2.23 | −2.44 | U | U | |
| Au12Pd1 | O2 | −0.37 | U | U | −0.63 | U |
| CO | −1.41 | −1.18 | U | U | U | |
| Au13 | O2 | −0.44 | U | U | U | U |
| CO | −1.36 | −1.29 | U | U | U | |
For O2 adsorption, we calculated five possible configurations (see Fig. 3): (1) on the top (T), (2) on the bridge (B), (3) on the hollow (H), (4) between two top sites (B1), and (5) between the hollow site and the top site (H–T). For O2 adsorption on B1 and H–T sites, the molecule prefers a “side-on” configuration with both oxygen atoms bound to the Au or Pd atoms.57 At the T, B, and H sites, however, O2 prefers an “end-on” configuration with one oxygen atom binding to the Pd atom. The corresponding adsorption energies are summarized in Table 2. The H–T configuration is the most stable O2 adsorption site on Pd13 and Pd12Au1 clusters, with adsorption energies of −1.43 and −1.58 eV, respectively. In contrast, the B1 site is the most stable O2 adsorption site for Au12Pd43, with an adsorption energy of −0.63 eV. For the Au13 cluster, the T site is the only stable configuration, with an adsorption energy of −0.44 eV.
The adsorption energies of CO at the most favourable adsorption site for the Pd13, Pd12Au1, Au12Pd1, and Au13 clusters are −2.44, −2.56, −1.41, and −1.36 eV, respectively. The adsorption energies of O2 at the most favourable adsorption site for the Pd13, Pd12Au1, Au12Pd1, and Au13 clusters are −1.43, −1.58, −0.63, and −0.44 eV, respectively. The adsorption strength of both CO and O2 follows the order Au13 < Au12Pd1 < Pd13 < Pd12Au1. To understand the differences in the adsorption strength among the four selected clusters, we investigated the electronic structures of these clusters by calculating their local density of states (LDOS; Fig. 4). The d orbitals of metals play an extremely important role in catalysing the oxidation of CO. To quantitatively investigate the changing trend of the cluster d orbitals in the reaction processes, we calculated the d-band centres of the clusters as:
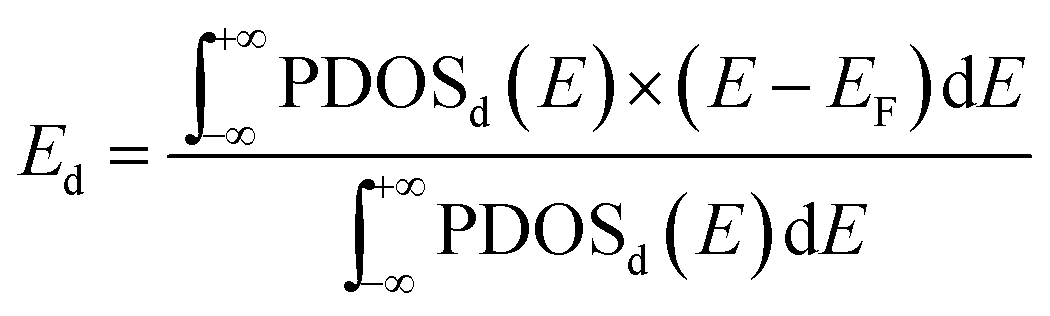 | (3) |
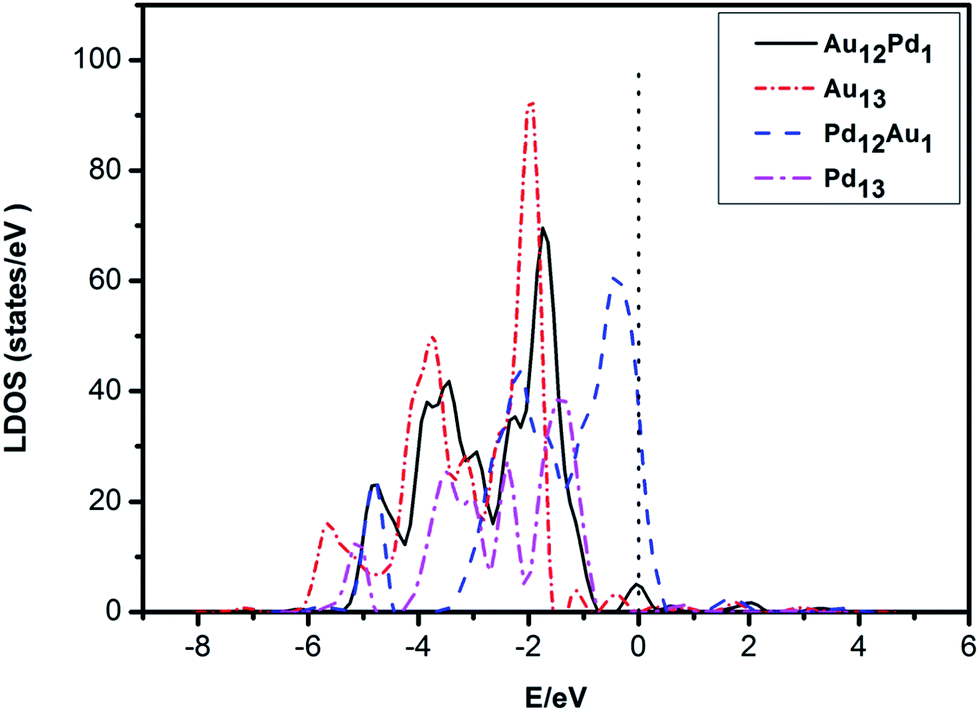 | ||
| Fig. 4 The electronic local density of states (LDOS) of the system projected on the d-states of icosahedral (ICO) Pd12Au1 and Pd13 clusters and truncated octahedral (TO) Au12Pd1 and Au13 clusters. | ||
The determined Ed of these clusters, which are listed in Table 1, follow the order: Au13 (−2.74 eV) < Au12Pd1 (−2.73 eV) < Pd13 (−2.28 eV) < Pd12Au1 (−1.51 eV). Accordingly, the more negative the d-band centre, the weaker the adsorption strength of CO and O2. This suggests that the position of the d-band centre is the determining factor for the adsorption strength of CO and O2.
We also find that the adsorption of CO on the clusters is much stronger than that of O2. This may be due to the stronger hybridization between the Au (Pd)-d and C-p states compared with the hybridization between the Au (Pd)-d and O-p states. As shown in Fig. 5A and B, the Au-d states for the Au12Pd1 cluster indicated by the peaks at −7 to −5 eV decrease upon CO and O2 adsorption due to electron depletion. This result supports the idea that the extent of the decrease in the electronic population of the d-orbital is larger for CO adsorption than for O2 adsorption. In addition, the Au-d states increase more obviously at the energy level from −12 to −7 eV for the Au12Pd1 cluster upon CO adsorption compared with O2 adsorption. This results from the electron accumulation and the increased hybridization region between the Au-d and C-p states. The charge density difference indicates a more distinct charge transfer phenomenon with CO adsorption (see Fig. 5B), again suggesting that the adsorption of CO on the clusters is much stronger than that of O2.
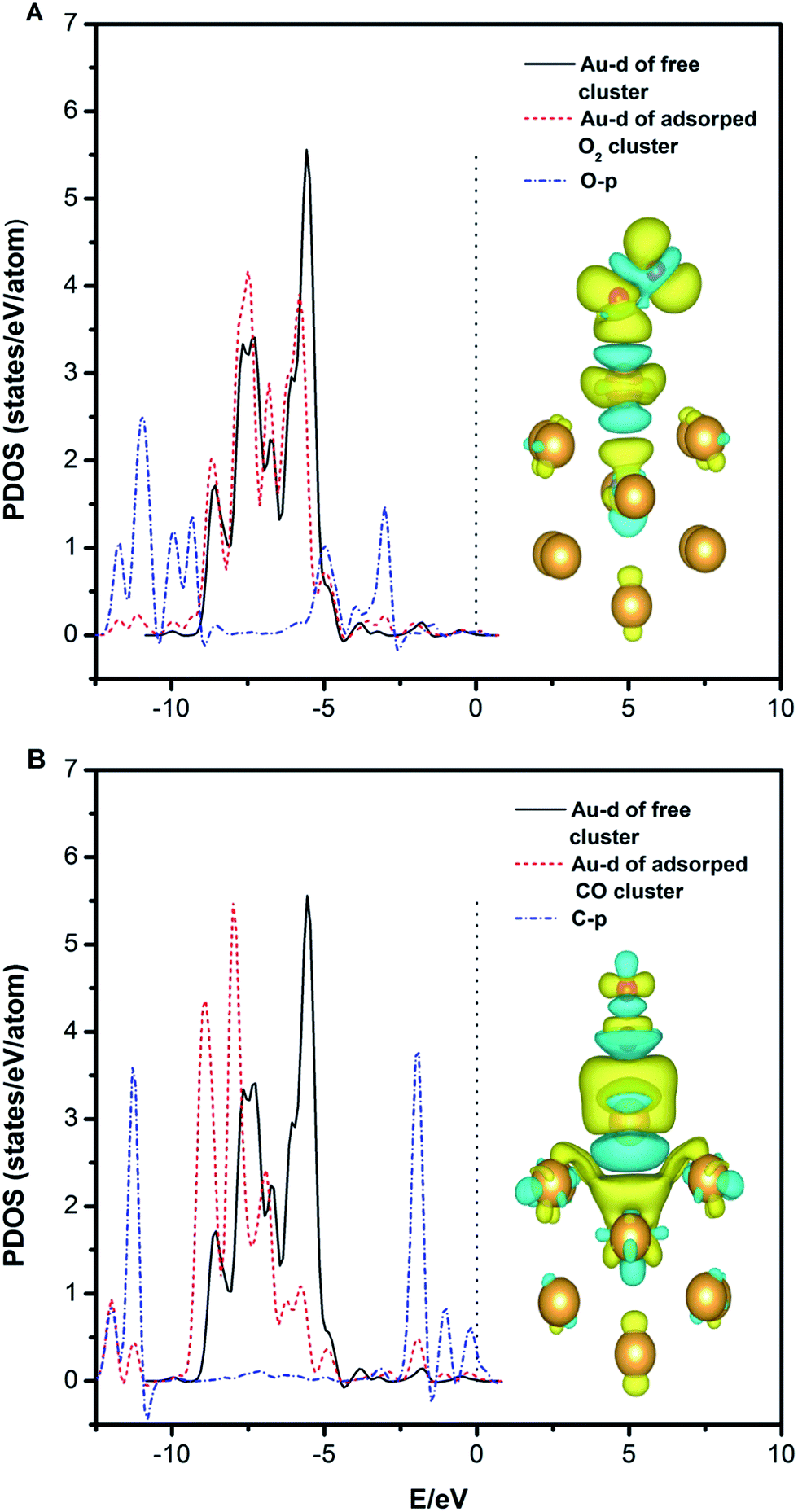 | ||
| Fig. 5 The partial density of states projected on the Au atom and the adatom (C or O) and the charge density difference plot of the free Au12Pd1 cluster upon O2 (A) and CO (B) adsorption. | ||
B. CO oxidation on free clusters
For CO oxidation, a bimolecular Langmuir–Hinshelwood (LH) mechanism is investigated since the adsorption of CO is much stronger than that of O2 on these clusters. The potential energy profiles and configurations for CO oxidation on Pd13, Pd12Au1, Au12Pd1, and Au13 clusters are shown in Fig. 6A–D, respectively. For clarity, the corresponding reaction energies and energy barriers for adsorption on these clusters are also listed in Table 3. To search for the minimum energy paths for CO oxidation, we select the most stable co-adsorption configuration as an initial state, where CO and O2 molecules are co-adsorbed at their most preferable sites according to the data in Table 2. For the Pd-rich clusters in this work, CO and O2 prefer to adsorb on the H and H–T sites of Pd13 and Pd12Au1, respectively; thus, the co-adsorption of the two molecules are too close. The choice of H (for CO) and the suboptimum B1 (for O2) is also too close. Alternatively, we absorb CO on the suboptimum site (B site) and O2 on the H–T site. For Au-rich clusters, CO and O2 are co-adsorbed at two T sites.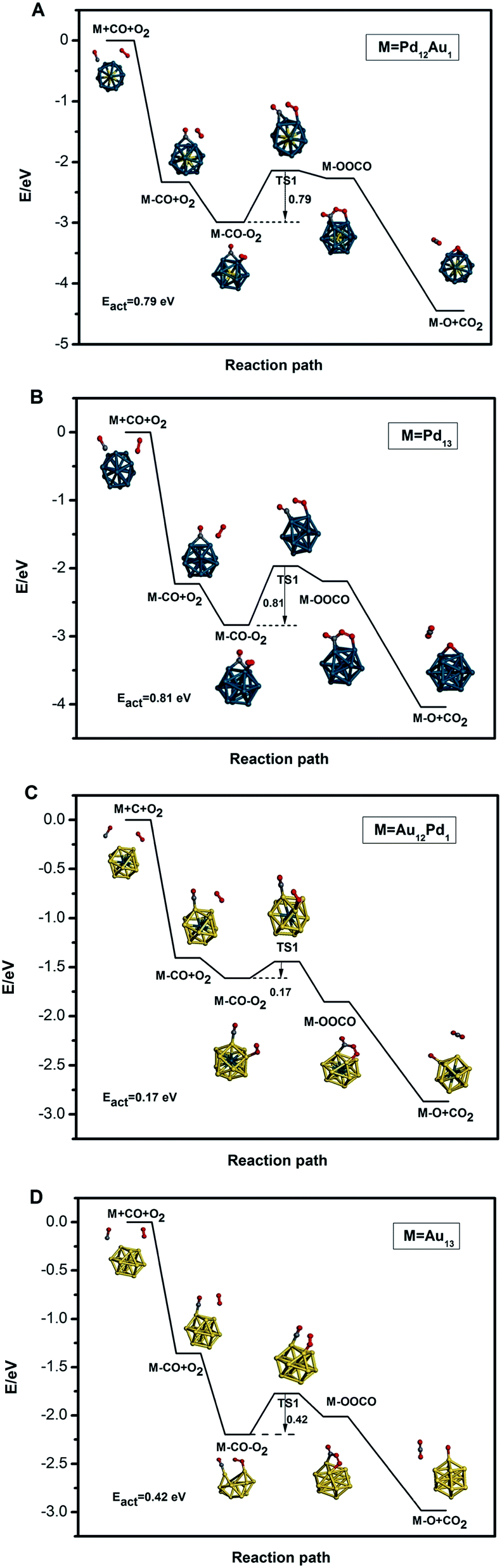 | ||
| Fig. 6 The minimum energy path (MEP) for CO adsorption, co-adsorption, oxidation, and desorption on the free Pd12Au1 (A), Pd13 (B), Au12Pd1 (C) and Au13 (D) clusters. The yellow, blue, grey, and red spheres represent Au, Pd, C, and O atoms, respectively. | ||
| Cluster | Adsorption energy | ||||
|---|---|---|---|---|---|
| Ead/CO | Ead/CO–O2 | Ead/OOCO | Ead/O+CO2 | Eact | |
| Pd12Au1 | −2.33 | −3.57 | −2.41 | −4.45 | 0.79 |
| Pd13 | −2.23 | −3.46 | −2.24 | −4.04 | 0.81 |
| Au13 | −1.36 | −3.12 | −2.09 | −2.98 | 0.42 |
| Au12Pd1 | −1.41 | −2.46 | −1.94 | −2.87 | 0.17 |
| Supported-Au12Pd1 | −1.12 | −1.76 | −1.68 | −3.20 | 0.41 |
Next, CO and O2 move closer to form the intermediate state OCOO* with a C–O single bond and a peroxide O–O bond, followed by the crossing of the first energy barrier (TS1). The corresponding energy barriers (TS1) are found to be 0.81, 0.79, 0.17, and 0.42 eV for Pd13, Pd12Au1, Au12Pd1, and Au13 clusters, respectively. Notably, the energy barriers for the formation of the intermediate state OCOO* (TS1) on Au-rich clusters are lower than those on the Pd-rich clusters. Among the free clusters, the Au12Pd1 cluster with TO structure exhibits the lowest energy barrier (0.17 eV) for CO oxidation.
In the third step, the peroxide O–O bond dissociates into two O atoms by crossing the second energy barrier (TS2). The calculated TS2 energy barriers are always zero for all the clusters considered in this work, meaning that CO2 is easily desorbed from the four free clusters without any barrier. To fully restore the catalytic system, the oxygen adatom left by the initially investigated reaction step needs to react with another CO molecule to produce another CO2 molecule. This often occurs as CO comes from the gas phase to form a second CO2 according to an Eley-Rideal (ER) mechanism; this reaction is generally barrier-less. One of the most important factors that can affect the reaction mechanism is the capture of oxygen. The low adsorption energies of O2 on CO-preadsorbed clusters (Ead(CO, O2) − Ead(CO)) significantly reduce the possibility of the CO + O2 reaction. In this work, the absolute values of Ead(CO, O2) − Ead(CO) are greater than 0.50 eV, enhancing the possibility of the CO + O2 reaction. From the analysis mentioned above, it is found that the Au12Pd1 cluster is the best candidate for CO oxidation among the free clusters studied herein.
C. CO oxidation on supported cluster
When the Au12Pd1 cluster is supported on single vacancy defective graphene, no structural change of the metal cluster is observed. To understand the structural stability of the supported metal cluster, the distortion energy (Edis) of the metal cluster is defined as the difference between the energy of the metal cluster in the configuration interacting with graphene and the energy of the bare cluster. The absolute value of Edis represents the distortion level; a negative value means that the cluster is changed into a more stable geometric configuration, while a positive value means that the cluster retains the original structure with relaxation upon adsorption. In this work, the Edis of the Au12Pd1 cluster is −0.46 eV, indicating an increase in stability when the Au12Pd1 cluster is supported on single vacancy defective graphene. In addition, the energy difference between the supported and free graphene is about −0.17 eV, indicating a slight structural change for the single vacancy defective graphene due to the addition of the Au12Pd1 cluster.The adsorption strengths of CO (−1.12 eV) and O2 (−0.30 eV) on the T sites of the single vacancy defective graphene-supported Au12Pd1 cluster are smaller than those on the corresponding free Au12Pd1 cluster at the same adsorption sites. This suggests that the monovacancy defective graphene support stably anchors the Au12Pd1 cluster at the vacancy site, consequently preventing the Au12Pd1 cluster from being significantly altered upon O2 or CO adsorption. To understand the difference in the adsorption strengths of the free and defective graphene-supported Au12Pd1 clusters, the LDOS of these clusters are calculated (Fig. 7A). Accordingly, the d-band centres are determined to be −2.73 and −2.81 eV for the free and defective graphene-supported Au12Pd1 clusters, respectively. These results agree with the point that the more negative the d-band centre, the weaker the adsorption strengths of CO and O2. In addition, a band gap of ∼0.5 eV between the highest occupied molecular orbital and the lowest unoccupied molecular orbital is found for the defective graphene-supported Au12Pd1 cluster, which can contribute to the enhanced stability58 (Fig. 7A).
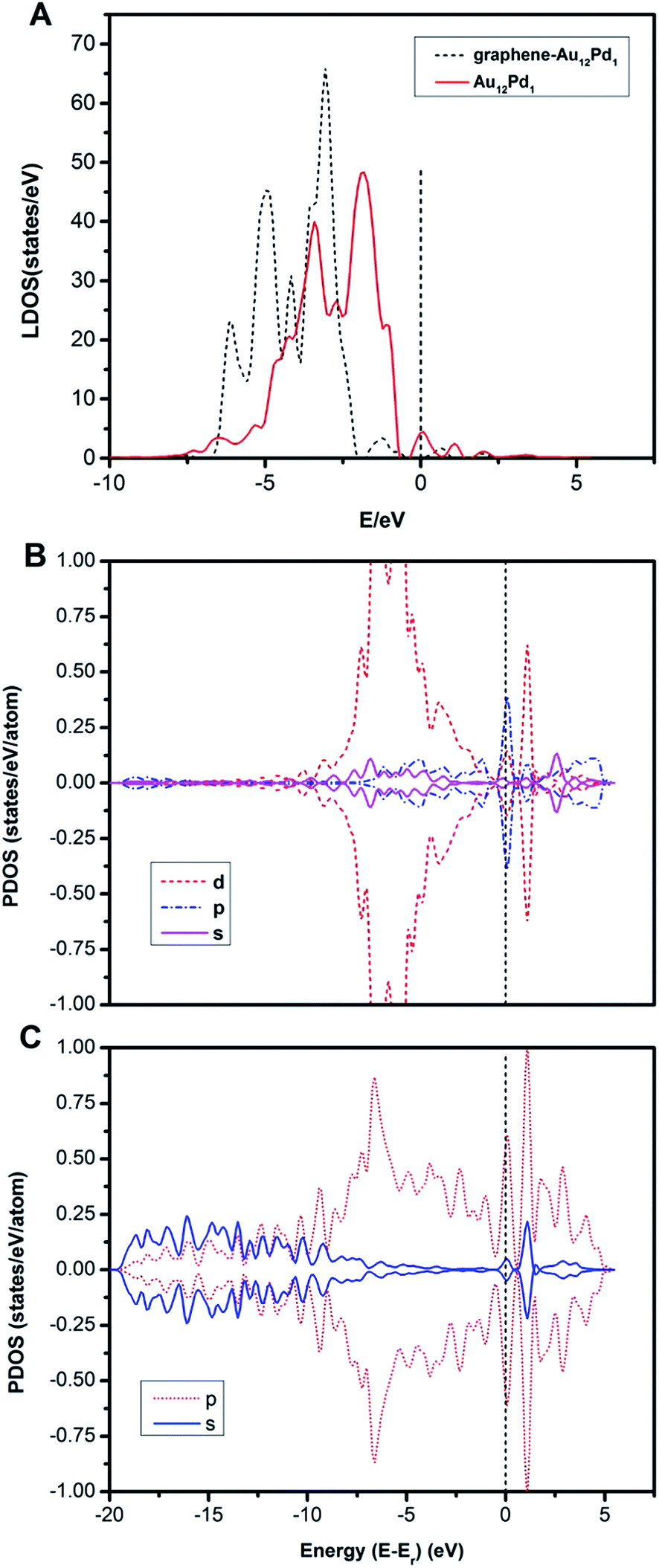 | ||
| Fig. 7 (A) LDOS of the system projected on the d-states of the free and graphene-supported Au12Pd1 clusters. (B) PDOS of the s, p and d states of bound Au atoms at the monovacancy site of graphene. (C) PDOS of the p and s states of carbon atoms nearest the monovacancy upon adsorption. Spin-up (↑) and spin-down (↓) states are marked as positive and negative values, respectively. | ||
The projected density of states (PDOS) has been analysed for the valence electrons of the defective graphene-supported Au12Pd1 cluster (Fig. 7B and C). As shown from the PDOS of the Au (binding at the monovacancy site of graphene) and C (nearest to the monovacancy) atoms, the strong hybridization between the Au-5d and C-2p states of the defective graphene occurs through almost the entire energy region, indicating a covalent bonding interaction between Au and C atoms. Note that the Au-5d orbital is important in the adsorption of reactants. The Löwdin charge analysis reveals that the negative excess charge on the bound Au atom is −0.20 e, and the total positive excess charge on the defective graphene is 0.18 e. This indicates that charge is transferred from the bound Au atom to defective graphene, possibly explaining the weaker O2 and CO adsorption energies on the defective graphene-supported Au12Pd1 system compared to the free Au12Pd1 cluster.59 Notably, the work-function can explain the charge transfer between the adsorbate and the substrate because both terms refer to energy required to remove electrons from an adsorbate-substrate system.59
Due to the larger adsorption strength of CO compared to O2, a bimolecular Langmuir–Hinshelwood (LH) mechanism is investigated for CO oxidation on the defective graphene-supported Au12Pd1 cluster. The adsorption energies and reaction energy barriers are listed in Table 3. The reaction path for CO catalytic oxidation on the defective graphene-supported Au12Pd1 cluster is also shown in Fig. 8. The energy barrier for the formation of the intermediate state OCOO* is 0.41 eV; this value is a little higher than that of the corresponding free cluster, but it is still a very low barrier (<0.5 eV). Notably, the energy barrier for CO oxidation on the Au12Pd1 cluster on graphene (0.42 eV) is lower than those for graphene-supported single Cu (0.54 eV),60 Si (0.57 eV),61 Fe (0.58 eV),26 and Pt (0.59 eV)27 atom catalysts. In conclusion, the single vacancy defective graphene-supported Au12Pd1 cluster exhibits an increase in stability, a decrease in the adsorption strength of CO and O2, and a slight increase in the energy barrier (0.41 eV) for CO oxidation compared to the corresponding free cluster.
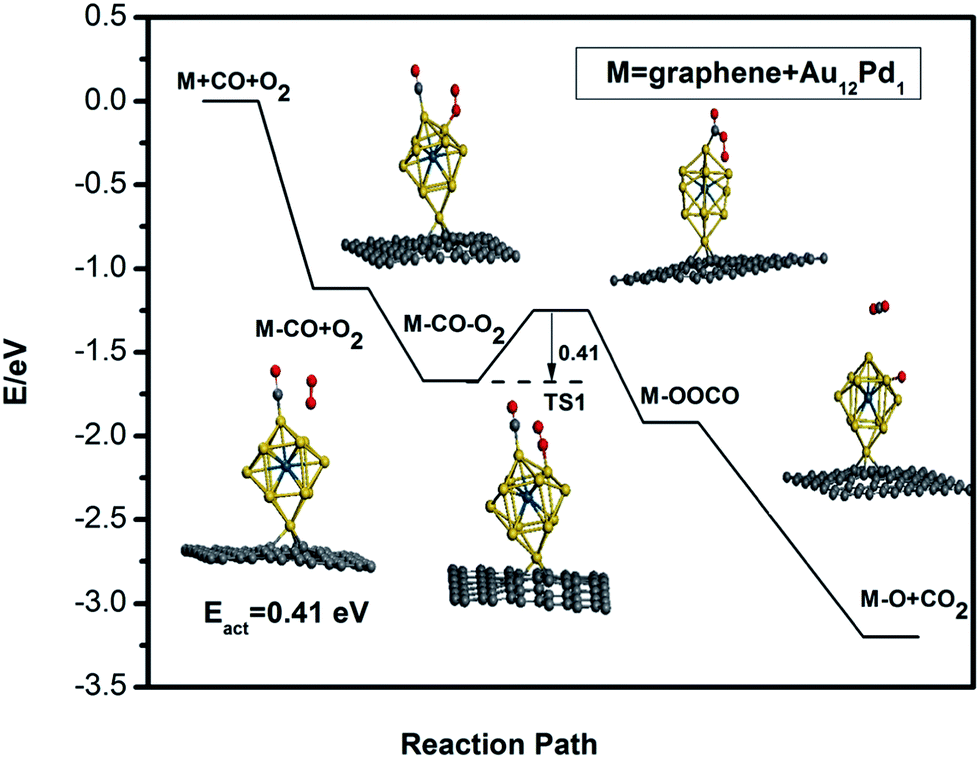 | ||
| Fig. 8 The minimum energy path (MEP) for CO adsorption, co-adsorption and oxidation on the defective graphene-supported Au12Pd1 cluster. | ||
IV. Conclusions
In summary, we investigated CO adsorption and oxidation on free and defective graphene-supported AumPdn (m + n = 13) clusters with either icosahedral (ICO) or truncated octahedral (TO) structures using density functional theory calculations. For the free AumPdn (m + n = 13) clusters, we studied the relatively stable structures of 13-atom Au–Pd clusters as well as the O2 and CO adsorption, the co-adsorption of CO and O2, and CO oxidation on these clusters. We found that the adsorption of CO is much stronger than that of O2 on the cluster surfaces, which is attributed to the strong hybridization between the metal-d and C-p orbitals. In addition, the structural transformation from ICO to TO is observed in the Au-rich clusters upon CO and O2 adsorption because the stability of the ICO cluster is lower than that of the TO structure. We also found that the free Au12Pd1 cluster with the TO structure exhibits the lowest energy barrier (0.17 eV) for CO oxidation among these free clusters. We comparatively studied the energy barriers for CO oxidation on the defective graphene-supported Au12Pd1 cluster and the corresponding free cluster. The results indicate that the single vacancy defective graphene-supported Au12Pd1 cluster possesses an increased stability, a decreased adsorption strength for CO and O2, and a slightly increased energy barrier for CO oxidation compared with the corresponding free cluster.Acknowledgements
This work is supported by the National Natural Science Foundation of China (21106003, 21176010, 91334203), the Beijing Novel Program (Z12111000250000), the “Chemical Grid Project” of BUCT and the Supercomputing Center of the Chinese Academy of Sciences (SCCAS).References
- Y. Z. Lu and W. Chen, Chem. Soc. Rev., 2012, 41, 3594 RSC.
- B. Nkosi, N. J. Coville and G. J. Hutching, J. Chem. Soc., Chem. Commun., 1988, 1, 71 RSC.
- M. Haruta, Nature, 2005, 437, 1098 CrossRef CAS PubMed.
- M. Haruta, T. Kobayashi, H. Sano and N. Yamada, Chem. Lett., 1987, 16, 405 CrossRef.
- A. S. K. Hashmi and G. J. Hutchings, Angew. Chem., Int. Ed., 2006, 45, 7896 CrossRef PubMed.
- Y. Gao, N. Shao, Y. Pei, Z. F. Chen and X. C. Zeng, ACS Nano, 2011, 5, 7818 CrossRef CAS PubMed.
- W. Liu, Y. F. Zhu and Q. Jiang, J. Phys. Chem. C, 2010, 114, 21094 CAS.
- O. Lopez-Acevedo, K. A. Kacprzak, J. Akola and H. Hakkinen, Nat. Chem., 2010, 2, 329 CrossRef CAS PubMed.
- B. Yoon, H. Hakkinen, U. Landman, A. S. Worz, J. M. Antonietti, S. Abbet, K. Judai and U. Heiz, Science, 2005, 307, 403 CrossRef CAS PubMed.
- B. Hammer and J. K. Norskov, Adv. Catal., 2000, 45, 71 CAS.
- X. Bokhimi, R. Zanella and C. Angeles-Chavez, J. Phys. Chem. C, 2010, 114, 14101 CAS.
- Y. Gao, N. Shao, Y. Pei and X. C. Zeng, Nano Lett., 2010, 10, 1055 CrossRef CAS PubMed.
- Y. Luo, H. O. Seo, K. D. Kim, M. J. Kim, W. S. Tai, M. Burkhart and Y. D. Kim, Catal. Lett., 2010, 134, 45 CrossRef CAS PubMed.
- J. Xu, T. White, P. Li, C. He, W. Y. Jianguo Yu and Y.-F. Han, J. Am. Chem. Soc., 2010, 132, 10398 CrossRef CAS PubMed.
- H. C. Ham, J. A. Stephens, G. S. Hwang, J. Han, S. W. Nam and T. H. Lim, J. Phys. Chem. Lett., 2012, 3, 566 CrossRef CAS.
- F. Gao, Y. L. Wang and D. W. Goodman, J. Phys. Chem. C, 2009, 113, 14993 CAS.
- K. Persson, A. Ersson, K. Jansson, N. Iverlund and S. Jaras, J. Catal., 2005, 231, 139 CrossRef CAS PubMed.
- J. B. Xu, T. S. Zhao, S. Y. Shen and Y. S. Li, Int. J. Hydrogen Energy, 2010, 35, 6490 CrossRef CAS PubMed.
- R. W. J. Scott, C. Sivadinarayana, O. Wilson and R. M. Crooks, J. Am. Chem. Soc., 2005, 127, 1380 CrossRef CAS PubMed.
- K. R. Ratinac, W. R. Yang, S. P. Ringer and F. Braet, Environ. Sci. Technol., 2010, 44, 1167 CrossRef CAS PubMed.
- A. K. Geim, Science, 2009, 324, 1530 CrossRef CAS PubMed.
- C. N. R. Rao, A. K. Sood, K. S. Subrahmanyam and A. Govindaraj, Angew. Chem., Int. Ed., 2009, 48, 7752 CrossRef CAS PubMed.
- A. Prestianni, A. Martorana, F. Labat, I. Ciofini and C. Adamo, J. Mol. Struct.: THEOCHEM, 2009, 903, 34 CrossRef CAS PubMed.
- Q. G. Jiang, Z. M. Ao, S. Li and Z. Wen, RSC Adv., 2014, 4, 20290 RSC.
- D. W. Boukhvalov and M. I. Katsnelson, J. Phys. Chem. C, 2009, 113, 14176 CAS.
- Y. F. Li, Z. Zhou, P. Jin, Y. S. Chen, S. B. B. Zhang and Z. F. Chen, J. Phys. Chem. C, 2010, 114, 6250 CAS.
- Y. Tang, Z. Yang and X. Dai, Phys. Chem. Chem. Phys., 2012, 14, 16566 RSC.
- D. H. Lim and J. Wilcox, J. Phys. Chem. C, 2011, 115, 22742 CAS.
- D. H. Lim, A. S. Negreira and J. Wilcox, J. Phys. Chem. C, 2011, 115, 8961 CAS.
- I. Fampiou and A. Ramasubramaniam, J. Phys. Chem. C, 2012, 116, 6543 CAS.
- A. J. Logsdail and J. Akola, J. Phys. Chem. C, 2011, 115, 15240 CAS.
- E. Aprà, F. Baletto, R. Ferrando and A. Fortunelli, Phys. Rev. Lett., 2004, 93, 065502 CrossRef.
- J. Zhang, H. M. Jin, M. B. Sullivan, F. Chiang, H. Lim and P. Wu, Phys. Chem. Chem. Phys., 2009, 11, 1441 RSC.
- D. W. Yuan, Z. R. Liu and J. H. Chen, J. Chem. Phys., 2011, 134, 054704 CrossRef CAS PubMed.
- H. L. Chen, C. H. Su and H. T. Chen, J. Chem. Phys., 2012, 536, 100 CAS.
- S. L. Peng, L. Y. Gan, R. Y. Tian and Y. J. Zhao, Comput. Theor. Chem., 2011, 977, 62 CrossRef CAS PubMed.
- B. Zhu, G. Thrimurthulu, L. Delannoy, C. Louis, C. Mottet, J. Creuze, B. Legrand and H. Guesmi, J. Catal., 2013, 308, 272 CrossRef CAS PubMed.
- E. Apra and A. Fortunelli, J. Phys. Chem. A, 2003, 107, 2934 CrossRef CAS.
- F. Baletto, R. Ferrando, A. Fortunelli, F. Montalenti and C. Mottet, J. Chem. Phys., 2002, 116, 3856 CrossRef CAS PubMed.
- D. Cheng, S. Huang and W. Wang, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 064117 CrossRef.
- D. Cheng, X. Liu, D. Cao, W. Wang and S. Huang, Nanotechnology, 2007, 18, 475702 CrossRef.
- D. Cheng and W. Wang, Nanoscale, 2012, 4, 2048 Search PubMed.
- R. A. Johnson, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 12554 CrossRef.
- R. A. Johnson, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 9717 CrossRef CAS.
- D. Cheng, I. S. Atanasov and M. Hou, Eur. Phys. J. D, 2011, 64, 37 CrossRef CAS.
- I. S. Atanasov and M. Hou, Eur. Phys. J. D, 2009, 52, 51 CrossRef CAS.
- I. Atanasov and M. Hou, Surf. Sci., 2009, 603, 2639 CrossRef CAS PubMed.
- P. Giannozzi, S. Baroni, N. Bonini, M. Calandra, R. Car, C. Cavazzoni, D. Ceresoli, G. L. Chiarotti, M. Cococcioni, I. Dabo, A. Dal Corso, S. de Gironcoli, S. Fabris, G. Fratesi, R. Gebauer, U. Gerstmann, C. Gougoussis, A. Kokalj, M. Lazzeri, L. Martin-Samos, N. Marzari, F. Mauri, R. Mazzarello, S. Paolini, A. Pasquarello, L. Paulatto, C. Sbraccia, S. Scandolo, G. Sclauzero, A. P. Seitsonen, A. Smogunov, P. Umari and R. M. Wentzcovitch, J. Phys.: Condens. Matter, 2009, 21, 359902 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 CrossRef CAS.
- D. Vanderbilt, Phys. Rev. B: Condens. Matter Mater. Phys., 1990, 41, 7892 CrossRef.
- K. Miwa and A. Fukumoto, Phys. Rev. B: Condens. Matter Mater. Phys., 2002, 65, 155114 CrossRef.
- B. S. Pujari, S. Gusarov, M. Brett and A. Kovalenko, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 84, 041402 CrossRef.
- D. Cheng, H. Xu and A. Fortunelli, J. Catal., 2014, 314, 47 CrossRef CAS PubMed.
- M. K. Srivastava, Y. Wang, A. F. Kemper and H. P. Cheng, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 165444 CrossRef.
- G. Henkelman, B. P. Uberuaga and H. Jonsson, J. Chem. Phys., 2000, 113, 9901 CrossRef CAS PubMed.
- Y. Xu, A. V. Ruban and M. Mavrikakis, J. Am. Chem. Soc., 2004, 126, 4717 CrossRef CAS PubMed.
- C. M. Chang, C. Cheng and C. M. Wei, J. Chem. Phys., 2008, 128, 124710 CrossRef CAS PubMed.
- G. Chen, S. J. Li, Y. Su, V. Wang, H. Mizuseki and Y. Kawazoe, J. Phys. Chem. C, 2011, 115, 20168 CAS.
- D. H. Lim and J. Wilcox, J. Phys. Chem. C, 2011, 115, 22742 CAS.
- E. H. Song, Z. Wen and Q. Jiang, J. Phys. Chem. C, 2011, 115, 3678 CAS.
- J. X. Zhao, Y. Chen and H. G. Fu, Theor. Chem. Acc., 2012, 131, 1 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |