
Layer-by-layer polyelectrolyte complex coated poly(methacrylic acid) nanogels as a drug delivery system for controlled release: structural effects†
Xiaorui Li,
Pengcheng Du and
Peng Liu*
State Key Laboratory of Applied Organic Chemistry and Institute of Polymer Science and Engineering, College of Chemistry and Chemical Engineering, Lanzhou University, Lanzhou 730000, China. E-mail: pliu@lzu.edu.cn; Fax: +86-931-8912582; Tel: +86-931-8912582
First published on 17th October 2014
Abstract
Core–shell microspheres have attracted intense interest as drug delivery system (DDS) because of their integrated advantages of the core and shell materials. Here the structural effects, such as the crosslinking degree of the cores and the thickness of the polyelectrolyte complex shells, on the dipyridamole (DIP) loading and release performance were investigated in detail for the first time with the core–shell poly(methacrylic acid)@(chitosan/alginate)n microsphere as a drug carrier model, fabricated by encapsulating poly(methacrylic acid) (PMAA) nanogels in the layer-by-layer (LbL) engineered chitosan/alginate (CS/AL) multilayer shells. The core–shell microspheres with two-bilayer chitosan/alginate shells (PMAA@(CS/AL)2) were selected for the in vitro controlled release of the water-insoluble anticancer drug (doxorubicin (DOX)) in simulated body fluids (SBF). After the encapsulation, the DOX-loading capacity increased from 32.18% to 40.12% and the effect of the media pH values on the cumulative release from the PMAA@(CS/AL)2 was more remarkable than the core material, indicating the encapsulation with the polyelectrolyte multilayer shells was favorable for the drug loading and pH-responsive controlled release. Furthermore, the polyelectrolyte multilayer shells also improved the cytocompatibility of the drug carriers. This understanding will lead to better design of smart core–shell DDS for controlled release.
1. Introduction
In recent years, core–shell microspheres with designed diameter and shell thickness have received increasing attention due to their integrated advantages in drug delivery.1 There are mainly two kinds of core–shell microspheres as DDS, classified based on the functions of the core and shell materials. In the first one, the hard core materials, such as magnetic nanoparticles or noble metal nanoparticles, have been encapsulated into the polymeric or porous silica shells to provide the magnetic-targeting, imaging or remote releasing, while the drugs could only be loaded into the shells.2–4 For the other one, both the core and shell materials could be used for the drug-loading, such as mesoporous silica or polymeric materials.5–8 In both kinds of the core–shell microspheres as DDS, the polymeric materials have been widely used as the shell materials due to their easy manipulation and stimuli-responsive controlled release characteristics, besides the drug-loading role similar as the mesoporous silica shells.The stimuli-responsive polymeric shells could be coated onto various cores via the in situ polymerization,3,6 microfluidic,9 or layer-by-layer (LbL) assembly technique.10,11 The LbL assembly technique, via stepwise deposition of opposite charged polyelectrolytes onto a core, has been acknowledged as a convenient and versatile method for the core–shell microspheres.12 Compared with other relevant techniques, the drug-loading capacity and the controlled release performance of the LbL engineered core–shell drug-carriers could be conveniently and precisely controlled by tailoring the assembly parameters such as layers of adsorption, ionic strength, and pH of dipping solution.13
As for the core materials, the polymeric cores might expand upon the external stimuli or drug-loading,14,15 whereas the volumes of the mesoporous inorganic cores remain constant under the same conditions. The volume expansion of the cores might be restricted by the polymeric shells in the core–shell drug-carriers. An efficient approach to overcome the disadvantage is to design the yolk–shell microspheres.16,17 Unfortunately, the synthetic procedure is complicated and only the crosslinked synthetic polymers could be used.
Up to now, many LbL engineered core–shell drug-carriers with responsive polymer nanogels as cores have been reported,18,19 but there is no reference on their structural effect. In the present work, the poly(methacrylic acid) (PMAA) nanogels with different crosslinking degrees were synthesized via the facile distillation–precipitation polymerization in acetonitrile with 2,2′-azodiisobutyronitrile (AIBN) as initiator and divinylbenzene (DVB45) as crosslinking reagent. In order to realize the better controlled releasing performance, the chitosan/alginate multilayer shells ((CS/AL)n) with different thicknesses were coated onto the PMAA nanogels by the LbL assembly technique via electrostatic interaction between the amino groups of CS and the carboxyl groups of AL (Scheme 1). The effect of the cross-linking degrees of the PMAA nanogels and the thicknesses of the polyelectrolyte multilayer shells of the core–shell PMAA@(CS/AL)n microspheres on their drug-loading and pH-responsive controlled release performance were optimized with dipyridamole (DIP) as a model drug. Finally, the optimized core–shell microspheres were used as DDS for the in vitro controlled release of the hydrophobic anticancer drug (doxorubicin (DOX)) in simulated body fluids (SBF).
 | ||
| Scheme 1 Schematic illustration of the drug loading and controlled release of the PMAA nanogels and the core–shell PMAA@(CS/AL)4 microspheres. | ||
2. Experimental
Materials and reagents
CS with viscosity-average molecular weight of 6.0 × 105 and deacetylation degree of 90% was purchased from Zhengjiang Yuhuan Marine Biotechnol. Co., Jiangsu, China. AL with viscosity-average molecular weight of 4.5 × 105 was obtained from Xudong Chem. Plant, Beijing, China. DIP was analytical reagent grade from J & K Chemical Ltd. Doxorubicin hydrochloride (DOX·HCl) was purchased from Beijing Huafeng United Technology Co. Ltd., Beijing, China.DVB-45 were purchased from Tianjin Guangfu Chemical Co. Ltd., Tianjin, China and used without further purification. Methacrylic acid (MAA) (Tianjin Chemical Co. Ltd., Tianjin, China) was distilled under vacuum before use. Acetonitrile (analytically pure) was provided by Tianjin no. 3 Chemical Plant, Tianjin, China. AIBN (Tianjin Guangfu Chemical Co. Ltd., Tianjin, China) was recrystallized from ethanol before use. Deionized water was used throughout.
Monodisperse PMAA nanogels with different crosslinking degrees
The monodisperse PMAA nanogels were synthesized via the distillation–precipitation polymerization technique.20 Typically, 1.36 mL MAA, 0.252 mL DVB45 (5 mol% relative to MAA) and 32 mg AIBN (2.3 wt% relative to MAA) were dissolved into 80 mL acetonitrile. After the solution was degassed for at least 5 min by nitrogen purging to remove the dissolved oxygen, the reaction mixture was heated from room temperature to the boiling state within 30 min. The initially homogeneous mixture became milky white after 15 min. The reaction was ended after 42 mL acetonitrile had been distilled from the reaction system.The PMAA nanogels were separated by centrifugation and washing thoroughly with acetonitrile and water and dried at 50 °C in a vacuum oven. The other PMAA nanogels with different crosslinking degrees were synthesized by the same procedure except for altering the DVB45 feeding ratios (10%, 15%, 20%, and 25%) for comparison.
Encapsulating PMAA with CS/AL multilayer shells
The encapsulation of the PMAA-10 nanogels with the polyelectrolyte multilayer shells with different thicknesses were conducted by the LbL assembly technique at pH 5.0: 100 mL acetic acid solution containing 0.50 g CS was added to 200 mL aqueous dispersion containing 2.0 g PMAA-10, and the mixture was stirred for 8 h to accomplish the adsorption of CS. Then the core–shell microspheres were centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm and washed with water for four times to remove the dissociated CS and obtain the chitosan coated PMAA nanogels (PMAA@CS1). 100 mL aqueous solution containing 0.50 g AL was added to 200 mL aqueous dispersion of the PMAA@CS1 and stirred for 8 h. The PMAA nanogels coated with one polyelectrolyte bilayer shell (PMAA@(CS/AL)1) were obtained. Then, CS and AL were alternately deposited for further three times onto the core–shell PMAA@(CS/AL)1 microspheres to obtain the PMAA microspheres encapsulated with the CS/AL multilayer shells with different thicknesses, the core–shell (PMAA@(CS/AL)2, PMAA@(CS/AL)3, and PMAA@(CS/AL)4) microspheres (Scheme 1).
000 rpm and washed with water for four times to remove the dissociated CS and obtain the chitosan coated PMAA nanogels (PMAA@CS1). 100 mL aqueous solution containing 0.50 g AL was added to 200 mL aqueous dispersion of the PMAA@CS1 and stirred for 8 h. The PMAA nanogels coated with one polyelectrolyte bilayer shell (PMAA@(CS/AL)1) were obtained. Then, CS and AL were alternately deposited for further three times onto the core–shell PMAA@(CS/AL)1 microspheres to obtain the PMAA microspheres encapsulated with the CS/AL multilayer shells with different thicknesses, the core–shell (PMAA@(CS/AL)2, PMAA@(CS/AL)3, and PMAA@(CS/AL)4) microspheres (Scheme 1).
Effects of crosslinking degrees of PMAA nanogels and thicknesses of (CS/AL) shells on drug-loading and release performance
The obtained PMAA nanogels (50.0 mg) with different cross-linking degrees (5, 10, 15, 20, or 25 mol%) and the core–shell microspheres with different polyelectrolyte complex shell thicknesses (PMAA@(CS/AL)n (n = 1, 2, 3, or 4)) were added into 5.0 mL 1.0 mg mL−1 DIP solution (pH 4.0) at 25 °C for the drug-loading, respectively. After being stirred for 3 days, the DIP-loaded nanogels and microspheres were centrifuged to remove the excess DIP. Then the drug concentrations in the supernatant solution were measured using a UV spectrophotometer at a wavelength of 284.80 nm. The drug-loading capacities of the nanogels and microspheres were calculated from the drug concentrations in solutions before and after loading.A 10 mL buffer solution containing 10 mg DIP-loaded PMAA nanogels or DIP-loaded PMAA@(CS/AL)n microspheres was transferred into dialysis tube with a weight cutoff 14000 and immersed into 150 mL buffer solution at pH 1.8 or 7.4 at 37 °C, respectively. 5.0 mL dialysates were taken out at certain time intervals and analyzed with UV-vis spectrometry to detect the release rate. Furthermore, 5.0 mL fresh buffer solution was added after each sampling to keep the total solution volume constant. The cumulative release is expressed as the percentage of the released drug over time.
DOX-loading and controlled release
The PMAA-10 nanogels or the PMAA@(CS/AL)2 microspheres (10.0 mg) was added into 3.0 mL 1.0 mg mL−1 DOX solution at pH 4.0 and stirred for 3 days at 25 °C, respectively. The DOX-loaded nanogels and microspheres were centrifuged to remove the excess DOX. Then the drug concentrations in the supernatants were measured using UV spectrophotometer at a wavelength of 233.00 nm. The drug-loading capacities of the nanogels and microspheres were calculated from the drug concentrations in solution before and after loading. The drug release was carried out at pH 5.0 or 7.4 at 37 °C, with the same procedure as DIP. The release kinetics were analyzed with the Higuchi21 and Korsmeyer–Peppas22 models:| Higuchi model: Mt = kt1/2 |
| Korsmeyer–Peppas model: Mt/M∞ = ktn |
Cytocompatibility assays
Sulforhodamine-B (SRB) assay was applied to estimate the cytocompatibility of the PMAA-10 nanogels and the core–shell PMAA@(CS/AL)2 microspheres with HepG2 cells. After the cells were seeded into 96-well plates at densities of 1 × 105 cells per well for 24 h, the drug carriers with different concentrations were added and incubated for 48 h. The cells were washed three times with phosphate buffered saline and the cell viability was measured by the SRB assay: the cells were fixed with the 50% trichloroacetic acid and stained with 0.4% SBR in 1% acetic acid. The cell-bound dye was extracted with 10 mL unbuffered Tris buffer solution, and measured at 550 nm using a plate reader.Analysis and characterization
The particle sizes and morphologies of the PMAA nanogels with different crosslinking degrees and the core–shell microspheres with different polyelectrolyte complex shell thicknesses (PMAA@(CS/AL)n (n = 1, 2, 3, or 4)) were characterized with a JEM1200 EX/S transmission electron microscope (TEM). The samples were dispersed in deionized water and stirred for 30 min, and then deposited on a copper grid covered with a perforated carbon film.The zeta potentials of the PMAA nanogels and the core–shell PMAA@(CS/AL)4 microspheres were determined with Zetasizer Nano ZS (Malven Instruments Ltd., UK), by adjusting the pH of sample solutions with NaOH or HCl solution.
The mean hydrodynamic diameters (Dh) of the PMAA nanogels and the core–shell PMAA@(CS/AL)n microspheres were determined by dynamical mode (dynamic light scattering (DLS)) on the “Light Scattering System BI-200SM, Brookhaven Instruments” device equipped with the BI-200SM Goniometer, the BI-9000AT Correlator, Temperature Controller and the Coherent INOVA 70C argon ion laser at 20 °C. DLS measurements are performed using 135 mW intense laser excitation at 514.5 nm and at a detection angle of 90° at 25 °C. The pH of sample solutions was adjusted with NaOH or HCl solution.
The drug loading and controlled release performance of the PMAA nanogels and the core–shell PMAA@(CS/AL)n microspheres were tracked by a Lambda 35 UV/vis spectrometer (Perkin-Elmer Instruments, USA) at room temperature.
3. Results and discussion
PMAA nanogels
The PMAA nanogels with different crosslinking degrees were synthesized by the distillation–precipitation polymerization. All the PMAA nanogels had unique spherical shape with smooth surface (Fig. 1). Their average diameters increased from 277.5 ± 6.1 to 301.0 ± 4.4, 358.0 ± 12.0, 463.3 ± 3.7, 492.8 ± 3.0 nm with increasing the DVB45 feeding ratio from 5 to 10, 15, 20, and 25 mol%, respectively. This might be due to that the crosslinked oligomers can easily precipitate from the solution onto the nuclei surface.18 | ||
| Fig. 1 TEM images of the PMAA nanogels prepared with different feeding ratios of DVB45: (a) 5 mol%, (b) 10 mol%, (c) 15 mol%, (d) 20 mol%, and (e) 25 mol%, and the core–shell PMAA@(CS/AL)n microspheres with different shell thicknesses (f) n = 1, (g) n = 2, (h) n = 3 and (i) n = 4. | ||
The mean hydrodynamic diameters (Dh) of the swollen PMAA nanogels (PMAA-5, PMAA-10, PMAA-15, PMAA-20, and PMAA-25) were 364.9 ± 4.9, 375.3 ± 5.8, 394.4 ± 8.7, 505.7 ± 11.7, and 522.5 ± 4.2 nm from DLS, respectively. Comparing the diameters determined by TEM and DLS, which represented the particle sizes in their dried and swollen states respectively, the volume swelling ratios (volume ratio of the swollen state and dried state) of the PMAA-5, PMAA-10, PMAA-15, PMAA-20, and PMAA-25 nanogels could be calculated to be 227.4%, 193.8%, 133.7%, 130.0%, and 119.2%, respectively. The PMAA-5 nanogels exhibited the most remarkable swelling behavior. It indicated that the swelling properties of the PMAA nanogels prepared with lower feeding ratios of crosslinker were better than those with higher ones, due to that the polymer chains are more rigid and the crosslinked network is more compact when higher feeding ratios of crosslinker are used. The higher crosslinking degree might result in the lower drug-loading capacity, due to the two main factors: the drug-loading space and the diffusion of the drug molecules into the hydrogels.23 Thus the PMAA-10 nanogels with a medium crosslinking degree were chosen for the further experiments.
The Dh of the PMAA-5, PMAA-10 and PMAA-15 nanogels gradually decreased from 395, 443 and 467 nm to 323, 336 and 349 nm with decreasing the pH value from 10.0 to 3.0, respectively (Fig. 2(A)). Evidently, the increased swelling at higher pH values was induced by ionization of the carboxylic groups in the nanogels.24 The protonation/deprotonation of the pendent carboxylic acid groups also influences the swelling properties of the weak acidic PMAA chains, which are hydrophobic at lower pH media and hydrophilic at higher pH media.25 At higher pH values, the PMAA chains are deprotonated, resulting in the swelling of the nanogels; at lower pH values, the PMAA chains are protonated and the nanogels are in their collapsed state. So the PMAA nanogels are rigid microspheres, showing the smaller diameter (Fig. 2(A)).
 | ||
| Fig. 2 pH dependence of the Dh of the PMAA nanogels with different crosslinking degrees (A) and the core–shell PMAA@(CS/AL)4 microspheres. | ||
The loaded drugs could be discharged by the shrinking of the nanogels when they are exposed to the acidic media, such as accumulation in the tumor sites from the normal tissues. So the phase transition induced by the protonation/deprotonation could be used as a pH-controlled switching for the pH-stimuli responsive controlled drug release.26
Layer-by-layer assembly of polyelectrolyte multilayer shells
The LbL assembly technique was used to encapsulate the PMAA-10 nanogels with the CS/AL multilayer shells. Initially, the polycation CS was adsorbed onto the PMAA-10 nanogel cores by the electrostatic interaction between the amino groups of CS and the carboxyl groups of the PMAA-10 cores. Next, the polyanion AL was added to the aqueous dispersion of the chitosan coated PMAA nanogels (PMAA@CS1) to achieve the LbL assembly between CS and AL. Repeating this cycle for four cycles, the core–shell microspheres with different shell thicknesses (PMAA@(CS/AL)n) (n = 1, 2, 3, 4) were obtained, respectively.The zeta potentials of the core–shell PMAA@(CS/AL)n microspheres were measured to track the LbL assembly (Fig. 3(A)). The half bilayer numbers correspond to the CS deposition and the whole bilayer numbers represent the AL adsorption, the zero layer number corresponds to the PMAA-10 nanogel cores with the zeta potential of −37.6 mV. After the first CS layer was deposited, the zeta potential was +28.0 mV, indicating that the deposition of CS could completely cover the surface of the PMAA-10 cores. Then the zeta potentials changed to −21.9 mV after the adsorption of AL over the PMAA@CS1. With increasing the adsorbed layers, the symmetrically alternating zeta potentials indicated that the polyelectrolyte multilayer shells had been successfully deposited on the PMAA-10 cores by the LbL assembly.27
 | ||
| Fig. 3 Zeta potentials (A) and the mean hydrodynamic diameters (B) of the PMAA-10 nanogels encapsulated by CS/AL multilayer shells with different thicknesses (PMAA@(CS/AL)n). | ||
The successful assembly could also be revealed by the TEM analysis (Fig. 1). Obviously, as the bilayer number of the polyelectrolyte multilayer shell increased, the average diameter of the (PMAA@(CS/AL)n (n = 1, 2, 3, 4)) microspheres increased gradually from 302.0 ± 4.4 nm of the PMAA-10 cores to 311.7 ± 4.2, 315.6 ± 3.4, 322.7 ± 3.6 and 334.9 ± 3.3 nm of the core–shell (PMAA@(CS/AL)1, PMAA@(CS/AL)2, PMAA@(CS/AL)3, and PMAA@(CS/AL)4) microspheres, respectively. It indicated that the thickness of the polyelectrolyte shells could be efficiently tuned by controlling the numbers of the CS/AL bilayer coated.
The mean hydrodynamic diameters (Dh) of the PMAA-10 cores and the core–shell PMAA@(CS/AL)1, PMAA@(CS/AL)2, PMAA@(CS/AL)3 and PMAA@(CS/AL)4 microspheres were 375.3, 415.7, 456.7, 479.8, and 498.2 nm, respectively (Fig. 3(B)). The Dh increased 40.4, 41.0, 23.1, and 18.4 nm while coating one, two, three, and four polyelectrolyte bilayers respectively, also indicating that the increased shell thickness with increasing the numbers of the polyelectrolyte bilayers coated. Their volume swelling ratios increased from 191.9% of the PMAA-10 cores to 237.2%, 303.0%, 328.7%, and 329.2% of the core–shell (PMAA@(CS/AL)n (n = 1, 2, 3, 4)) microspheres, respectively. It meant that the polyelectrolyte multilayer shells exhibited the higher swelling ability due to their non-covalent interlayer interaction. And the swelling ability tended to be steady with the CS/AL bilayer number more than 2. It demonstrated that the less additional polyelectrolyte could be adsorbed with more polyelectrolyte complex layers coated, as shown as the decreased amplitude in the zeta potential results (Fig. 3(A)).
The zeta potentials of the PMAA-10 nanogels and the core–shell PMAA@(CS/AL)4 microspheres at media with different pH values are compared in Fig. 4. In the whole range, the zeta potentials of the PMAA-10 nanogels were negative and their absolute values increased with increasing the pH values, due to that more neutral carboxylic acid groups of the PMAA nanogels deprotonated into the negative carboxylate ions in the higher pH media. As for the core–shell PMAA@(CS/AL)4 microspheres, their zeta potentials changed from positive to negative near pH 5, indicating that the core–shell PMAA@(CS/AL)4 microspheres have pH-dependent charge-conversional feature, which might be favor to the cellular internalization in the acidic tumor extracellular environments (as acidic as pH 5.7).28 Thus, their negatively charged surface in the neutral or basic media can maintain their stealth character during blood circulation, spontaneously and precisely trigger responsive drug release or enhance interaction between nanocarriers and targeting cells at the targeting tumor sites.
 | ||
| Fig. 4 Zeta potentials of the PMAA-10 nanogels and the core–shell PMAA@(CS/AL)4 microspheres at different pH values. | ||
The zeta potential gap between the PMAA-10 nanogels and the core–shell PMAA@(CS/AL)4 microspheres was 53.8 mV at pH 3.0. As the media pH values increased, the gaps decreased rapidly to only 0.3 mV at pH 8.0 due to the polyelectrolyte multilayer shells coated. It could be ascribed that the CS/AL could form a dense shell to hide the influence of the charges from the surface beneath.29 It showed that the polyelectrolyte multilayer shells might act as a switch to tune the drug release from the PMAA cores.
pH stimuli-responsive characteristics of the core–shell PMAA@(CS/AL)4 microspheres
The influence of the media pH values on the Dh of the core–shell PMAA@(CS/AL)4 microspheres was investigated by DLS. It decreased from 592 nm to 376 nm with increasing pH values in the range of 3 to 11 (Fig. 2(B)). It is well known that the pKa values of the guluronic acid and the mannuronic acid of AL are 3.65 and 3.38, respectively.30 However, CS is a weak polybase with pKa value of about 6.5,31 and the charge densities of AL and CS are mainly determined by the media pH values. At lower pH (<4), the ionization of the carboxyl groups was normally depressed. The size of the core–shell PMAA@(CS/AL)4 microspheres increased more significantly because the decreasing pH values weakened the electrostatic interaction between AL and CS in the polyelectrolyte multilayer shells, although the PMAA cores shrunk. In the pH range of 4–7, the size decreasing extent of the core–shell PMAA@(CS/AL)4 microspheres was smaller compared to pH < 4 and pH > 7 because of the significant electrostatic attraction between AL and CS. The ionization of the amino groups of CS decreases greatly when the media pH values increases above 6.0 (around the pKa of chitosan 6.5), while less than 10% amino groups is usually ionized at pH values higher than 7.5.32 The decrease in size might be due to the insolubility and shrinkage of CS in the basic media. The shrinking degree of the core–shell PMAA@(CS/AL)4 microspheres with increasing media pH values was slightly lower than those hollow CS/AL multilayered microspheres reported previously,34 due to the remarkable swelling of the PMAA-10 nanogels in the basic media (Fig. 2(A)).Effects of crosslinking degrees of PMAA nanogels and polyelectrolyte multilayer thicknesses on drug loading and release performance
DIP was loaded onto the PMAA nanogels in aqueous solution at pH 4.0 at 25 °C. The drug-loading capacities of the PMAA nanogels were 82.68, 45.30, 36.32, 24.70, and 18.38 mg g−1 for the PMAA-5, PMAA-10, PMAA-15, PMAA-20, and PMAA-25 nanogels, respectively. With increasing the feeding ratios of the crosslinker, the drug-loading capacities decreased significantly. The polymer chains are more flexible with a low feeding ratio of the crosslinking reagent, the mash is larger and elastic or the retractable drawstrings are weaker than those with higher feeding ratios, so it could load much more drug molecules under the same conditions.As for the core–shell PMAA@(CS/AL)n microspheres, the drug-loading capacities at pH 4.0 at 25 °C were 58.76, 55.26, 51.28, and 49.78 mg g−1 for the PMAA@(CS/AL)1, PMAA@(CS/AL)2, PMAA@(CS/AL)3 and PMAA@(CS/AL)4, respectively. All the data were higher than that of the PMAA-10 nanogels (45.30 mg g−1) under the same condition, indicating that the encapsulation with the polyelectrolyte complex shells could enhance the drug-loading capacity due to the electrostatic interaction between the amino group-containing drugs and the carboxylic acid groups of AL,33 as well as the hydrogen bonds between the drug and the polyelectrolytes. However, the drug-loading capacities decreased with increasing the thickness of the polyelectrolyte complex shells. The coated polyelectrolyte shells might have two opposite effects on the drug-loading. The drug-loading capacity could be enhanced via the interaction between drug and the polyelectrolytes via the electrostatic interaction or hydrogen bonds. But also, contrastingly, the volume expansion of the PMAA nanogels during the drug-loading is suppressed by the shells coated, thus the drug-loading might be hindered. The combination of these two effects results in the decline in drug-loading capacity with increasing the thickness of the polyelectrolyte shells.
The cumulative DIP release from the DIP-loaded PMAA nanogels (PMAA-5, PMAA-10, and PMAA-15) were 61.97%, 42.97% and 30.50% at pH 1.8 and 55.11%, 25.83% and 17.23% at pH 7.4 at 37 °C, respectively (Fig. 5(A)). The faster releasing of DIP from all the three DIP-loaded PMAA nanogels were achieved at pH 1.8 due to the shrinkage of the PMAA nanogels and the dissolution of DIP in acidic media. It was also found that the cumulative DIP release decreased with increasing their crosslinking degree in the same releasing condition.
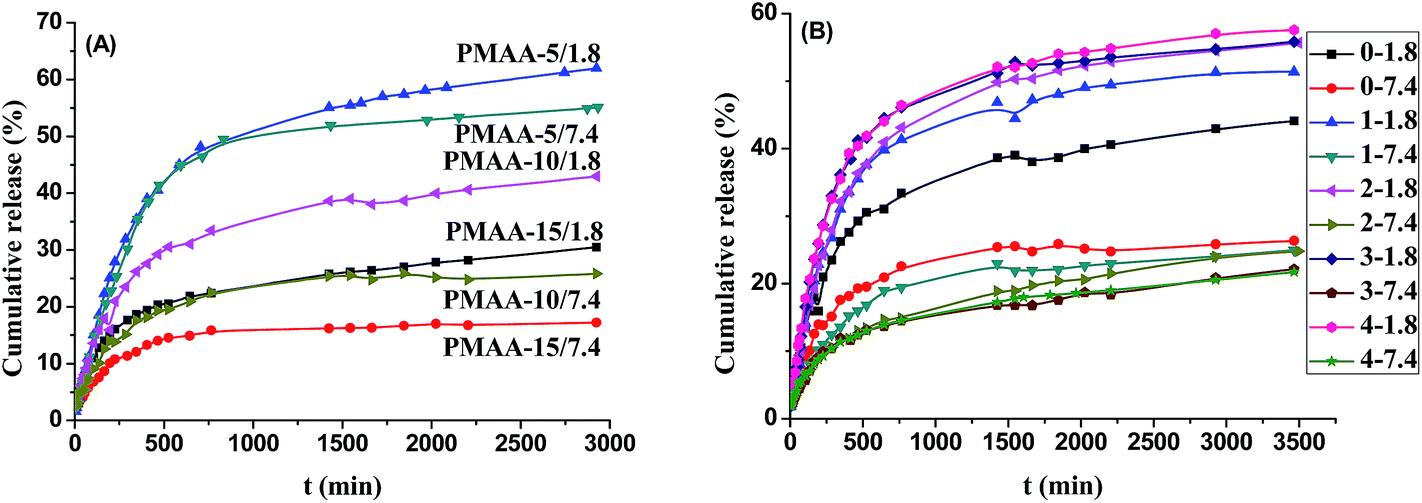 | ||
| Fig. 5 Cumulative dipyridamole (DIP) release from the DIP-loaded PMAA nanogels with different cross-linking degree in pH 1.8 and pH 7.4 buffer media at 37 °C (A), and from the core–shell PMAA@(CS/AL)n (n = 0, 1, 2, 3, 4) microspheres in pH 1.8 and pH 7.4 buffer media at 37 °C (B). The curves are denoted as the number of polyelectrolyte bilayer (n) and the pH value of the releasing media as n-pH value. | ||
More than 80% of the total cumulative DIP release was finished within 750 min for all the DDS regardless of the crosslinking degrees of the nanogels and the pH values of the releasing media. However, the long-term releasing rates were obviously faster in the acidic media than in the basic media. The drug releasing rates and the cumulative release from the DIP-loaded PMAA-5 nanogels within 750 min were similar in both pH 1.8 and pH 7.4 media, indicating that the most drug release (about 80% of the total cumulative DIP release) depended on the shrinkage of the PMAA nanogels with low crosslinking degree, while the long-term releasing was resulted from the dissolution of the drug loaded. As the crosslinking degrees of the PMAA nanogels increased to 10% and 15% of the PMAA-10 and PMAA-15 nanogels, the cumulative release within 750 min in basic media was only about 70% of those in the acidic media. It indicated that the cumulative release had been mainly affected by the shrinkage of the PMAA nanogels and the diffusion of the drug molecules through the nanogels.34
The time dependence of the cumulative DIP release from the drug-loaded PMAA@(CS/AL)n (n = 0, 1, 2, 3, 4) microspheres at pH 1.8 and 7.4 at 37 °C are compared in Fig. 5(B). Different from the releasing performance of the PMAA nanogels, only about 70% of the total cumulative DIP release was finished within 750 min for all the PMAA@(CS/AL)n microspheres regardless of the thicknesses of the polyelectrolyte multilayer shells (CS/AL)n (n = 1, 2, 3, 4) and the pH values of the releasing media. It indicated that the polyelectrolyte (CS/AL)n multilayer shells could slow down the releasing rate of the PMAA-10 nanogels. It was also found that the release rate and the cumulative DIP release from the drug carriers at pH 1.8 were faster and higher than those at pH 7.4 at the same temperature. It could be speculated that the lower release rate might be ascribed to the DIP solubility in different pH media. DIP became insoluble at high pH values because it is an alkaline molecule which could only be dissolved in acidic media. That is to say, the difference in release rates also should be partly attributed to the solubility of the drugs in different pH media.
The cumulative DIP release increased with the increasing of the thicknesses of the polyelectrolyte multilayer shells (CS/AL)n at pH 1.8 due to the fast release of the drug loaded onto the polyelectrolyte multilayer shells, while the cumulative DIP release decreased with increasing the thicknesses of the polyelectrolyte multilayer shells (CS/AL)n in pH 7.4 media. In other words, the polyelectrolyte multilayer (CS/AL)n (n = 1, 2, 3, 4) shells could accelerate the drug release in the acidic media but retard it in the basic media. It indicated that the polyelectrolyte multilayer (CS/AL)n shells were favorite to the pH-responsive controlled release of drugs from the core–shell drug carriers. As shown in Fig. 2(B), the size of the core–shell PMAA@(CS/AL)4 microspheres shrank significantly with increasing the media pH values. The drug molecules were difficult to diffuse throughout the polyelectrolyte (CS/AL)4 shells. Furthermore, the difference between the drug release from the drug carriers at pH 1.8 and 7.4 decreased with increasing the thicknesses of the polyelectrolyte (CS/AL)n shells. Compromising on the drug-loading capacity and the controlled release performance, the two polyelectrolyte bilayer (CS/AL)2 was selected for the core–shell drug carriers.
DOX-loading and controlled release
The DOX-loading capacities of the PMAA-10 nanogels and the core–shell PMAA@(CS/AL)2 microspheres at pH 4.0 at 25 °C were 32.18%, and 40.12%, respectively. The DOX-loading capacity was enhanced by encapsulation with the CS/AL bilayer shells, similar as the DIP-loading above-mentioned. It was resulted from the interaction between the polyelectrolytes and the drug, although the polyelectrolyte complex shells restricted the expansion of the PMAA-10 nanogels during the drug-loading.Due to the acidic intracellular microenvironments such as inside endosomes and lysosomes (pH 4.5–6.5) after nonspecific adsorptive endocytosis,35 the DOX release was compared in the simulated normal tissue (pH 7.4) and the simulated tumor intracellular microenvironments (pH 5.0). The pH-dependent DOX release was observed that the cumulative DOX release from DOX-loaded PMAA@(CS/AL)2 core–shell microspheres at pH 5.0 (52.44% and 72.14%) were higher than those at pH 7.4 (24.91% and 21.31%) within 50 h (Fig. 6). The cumulative DOX release from the PMAA@(CS/AL)2 microspheres at pH 5.0 was faster than at pH 7.4, partly due to that the permeability of the polyelectrolytes complex shell is better at lower pH values than at higher pH values, because that the decreasing pH values weaken the interaction between CS and AL,36 so that the Dh of the core–shell PMAA@(CS/AL)4 microspheres increased from 376 nm to 592 nm with decreasing pH values from 11 to 3, as shown in Fig. 2(B). Additionally, the acid-soluble nature of DOX might be another reason. The pH-dependent DOX release has a great advantage in curing the cancer cells with acidic microenvironments, especially with the polyelectrolyte multilayer shells.
 | ||
| Fig. 6 Cumulative DOX release from the DOX-loaded PMAA-10 and the DOX-loaded PMAA@(CS/AL)2 core–shell microspheres at pH 5.0 and pH 7.4 at 37 °C. | ||
The release data were analyzed on the basis of the Korsmeyer–Peppas equation and Higuchi kinetics. The coefficients of correlation (R2) of the Korsmeyer–Peppas equations were better than that of the Higuchi equation, and the n values of the Korsmeyer–Peppas equations were less than 0.5 (Fig. 1S†), so it could be concluded that the DOX releasing mechanism from the DOX-loaded PMAA-10 nanogels or the DOX-loaded PMAA@(CS/AL)2 microspheres at pH 5.0 and 7.4 should follow the non-Fickian model.37
Cytocompatibility assays
The in vitro cytocompatibility of the PMAA-10 cores and the core–shell PMAA@(CS/AL)2 microspheres were carried out with HepG2 cells using SRB assays. The cells were incubated with the drug carriers for 48 h at various concentrations (0–40 μg mL−1). The relative cell viability (ratio of cell viabilities of the testing sample with the control sample) of the core–shell PMAA@(CS/AL)2 microspheres was higher than that with the PMAA-10 cores up to 40 μg mL−1 (Fig. 7). It indicated that the (CS/AL)2 shells could also provide the drug-carrier better cytocompatibility.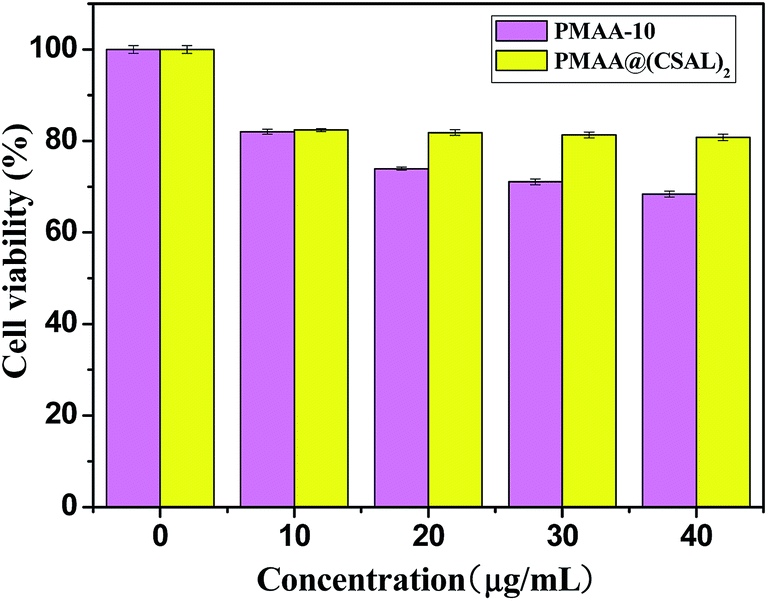 | ||
| Fig. 7 Cell viability data of the PMAA-10 cores and the core–shell PMAA@(CS/AL)2 microspheres. | ||
4. Conclusions
Aimed on the better design of smart drug delivery system (DDS) for controlled release, the core–shell poly(methacrylic acid)@(chitosan/alginate)n microspheres (PMAA@(CS/AL)n) were designed as a model DDS to understand the structural impact on drug-loading capacity and stimuli-responsive controlled release performance, such as the crosslinking degrees of the PMAA nanogels and the polyelectrolyte complex shell thicknesses of the core–shell (PMAA@(CS/AL)n) microspheres. The PMAA nanogels with a lower crosslinking degree were found to be the best core candidate and two-bilayer of the chitosan/alginate ((CS/AL)2) was the most effective polyelectrolyte complex shells. It was found that the CS/AL multilayer shells could enhance the drug-loading capacity, play a role of pH-controlled switching for the pH-responsive controlled release, and provide the drug-carrier better biocompatibility and the pH-dependent charge-conversional features. These results implied that the core–shell (PMAA@(CS/AL)n) microspheres are promising platform to construct pH-responsive controlled drug delivery system.Acknowledgements
This project was granted financial support from the National Nature Science Foundation of China (Grant no. 20904017) and the Program for New Century Excellent Talents in University (Grant no. NCET-09-0441).References
- I. d'Angelo, C. Conte, A. Miro, F. Quaglia and F. Ungaro, Expert Opin. Drug Delivery, 2014, 11, 283 CrossRef PubMed.
- M. Liong, J. Lu, M. Kovochich, T. Xia, S. G. Ruehm, A. E. Nel, F. Tamanoi and J. I. Zink, ACS Nano, 2008, 2, 889 CrossRef CAS PubMed.
- J. Kim, H. S. Kim, N. Lee, T. Kim, H. Kim, T. Yu, I. C. Song, W. K. Moon and T. Hyeon, Angew. Chem., Int. Ed., 2008, 47, 8438 CrossRef CAS PubMed.
- C. R. Thomas, D. P. Ferris, J. H. Lee, E. Choi, M. H. Cho, E. S. Kim, J. F. Stoddart, J. S. Shin, J. Cheon and J. I. Zink, J. Am. Chem. Soc., 2010, 132, 10623 CrossRef CAS PubMed.
- D. C. Niu, Z. Ma, Y. S. Li and J. L. Shi, J. Am. Chem. Soc., 2010, 132, 15144 CrossRef CAS PubMed.
- X. J. Kang, Z. Y. Cheng, D. M. Yang, P. A. Ma, M. M. Shang, C. Peng, Y. L. Dai and J. Lin, Adv. Funct. Mater., 2012, 22, 1470 CrossRef CAS.
- C. T. Wu, W. Fan, M. Gelinsky, Y. Xiao, J. Chang, T. Friis and G. Cuniberti, J. R. Soc., Interface, 2011, 8, 1804 CrossRef CAS PubMed.
- Y. J. Wang, Y. C. Chien, C. H. Wu and D. M. Liu, Mol. Pharmaceutics, 2011, 8, 2339 CrossRef CAS PubMed.
- J. Wu, T. T. Kong, K. W. K. Yeung, H. C. Shum, K. M. C. Cheung, L. Q. Wang and M. K. T. To, Acta Biomater., 2013, 9, 7410 CrossRef CAS PubMed.
- Y. Zhu, J. Shi, W. Shen, X. Dong, J. Feng, M. Ruan and Y. Li, Angew. Chem., 2005, 117, 5213 CrossRef.
- T. G. Shutava, S. S. Balkundi, P. Vangala, J. J. Steffan, R. L. Bigelow, J. A. Cardelli, D. Patrick O'Neal and Y. M. Lvov, ACS Nano, 2009, 3, 1877 CrossRef CAS PubMed.
- G. K. Such, A. P. R. Johnston and F. Caruso, Chem. Soc. Rev., 2011, 40, 19 RSC.
- Z. Tang, Y. Wang, P. Podsiadlo and N. A. Kotov, Adv. Mater., 2006, 18, 3203 CrossRef CAS.
- S. R. Deka, A. Quarta, R. D. Corato, A. Falqui, L. Manna, R. Cingolani and T. Pellegrino, Langmuir, 2010, 26, 10315 CrossRef CAS PubMed.
- Z. Y. Qiao, R. Zhang, F. S. Du, D. H. Liang and Z. C. Li, J. Controlled Release, 2011, 152, 57 CrossRef CAS PubMed.
- J. Liu, S. Z. Qiao, S. B. Hartono and G. Q. Lu, Angew. Chem., 2010, 122, 5101 CrossRef.
- P. C. Du, H. Y. Yang, J. Zeng and P. Liu, J. Mater. Chem. B, 2013, 1, 5298 RSC.
- R. Agrawal, A. Shanavas, S. Yadas, M. Aslam, D. Bahadur and R. Srivastava, J. Biomed. Nanotechnol., 2012, 8, 19 CrossRef CAS PubMed.
- G. Q. Fan, W. J. Tong, X. H. Hu and C. Y. Gao, Chem. J. Chin. Univ., 2008, 29, 2086 CAS.
- F. Bai, B. Huang, X. L. Yang and W. Q. Huang, Eur. Polym. J., 2007, 43, 3923 CrossRef CAS PubMed.
- T. Higuchi, J. Pharm. Sci., 1961, 50, 874 CrossRef CAS.
- R. W. Korsmeyer, R. Gurny, E. M. Doelker, P. Buri and N. A. Peppas, Int. J. Pharm., 1983, 15, 25 CrossRef CAS.
- P. Caliceti, S. Salmaso, A. Lante, M. Yoshida, R. Katakai, F. Martellini, L. H. I. Mei and M. Carenza, J. Controlled Release, 2001, 75, 173 CrossRef CAS.
- J. O. Kim, A. V. Kabanov and T. K. Bronich, J. Controlled Release, 2009, 138, 197 CrossRef CAS PubMed.
- N. Schuwer and H. A. Klok, Langmuir, 2011, 27, 4789 CrossRef CAS PubMed.
- J. X. Ding, X. L. Zhuang, C. S. Xiao, Y. L. Cheng, L. Zhao, C. L. He, Z. H. Tang and X. S. Chen, J. Mater. Chem., 2011, 21, 11383 RSC.
- M. K. Park, S. X. Deng and R. C. Advincula, Langmuir, 2005, 21, 5272 CrossRef CAS PubMed.
- R. van Sluis, Z. M. Bhujwalla, N. Raghunand, P. Ballesteros, J. Alvarez, S. Cerdan, J. P. Galons and R. J. Gillies, Magn. Reson. Med., 1999, 41, 743 CrossRef CAS.
- J. Zhou, G. Romero, E. Rojas, L. Ma, S. Moya and C. Y. Gao, J. Colloid Interface Sci., 2010, 345, 241 CrossRef CAS PubMed.
- G. Orive, S. Ponce, R. M. Hernandez, A. R. Gascon, M. Igartua and J. L. Pedraz, Biomaterials, 2002, 23, 3825 CrossRef CAS.
- J. L. Drury and D. J. Mooney, Biomaterials, 2003, 24, 4337 CrossRef CAS.
- M. Yalpani and L. D. Hall, Macromolecules, 1984, 17, 272 CrossRef CAS.
- B. Mu, P. Liu, Y. Dong, C. Y. Lu and X. L. Wu, J. Polym. Sci., Part A: Polym. Chem., 2010, 48, 3135 CrossRef CAS.
- Y. F. Shi, H. L. Lv, X. F. Lu, Y. Zhang and W. Xue, J. Mater. Chem., 2012, 22, 3889 RSC.
- R. F. Murphy, S. Powers and C. R. Cantor, J. Cell Biol., 1984, 98, 1757 CrossRef CAS.
- S. E. Burke and C. J. Barrett, Macromolecules, 2004, 37, 5375 CrossRef CAS.
- A. Raval, J. Parikh and C. Engineer, Ind. Eng. Chem. Res., 2011, 50, 9539 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra05066e |
| This journal is © The Royal Society of Chemistry 2014 |