
Differentially cross-linkable core–shell nanofibers for tunable delivery of anticancer drugs: synthesis, characterization and their anticancer efficacy†
Abstract
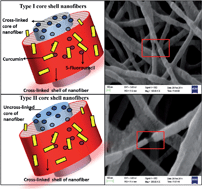
This work introduces a new dimension for controlled drug delivery by nanofiber based scaffolds for anticancer therapy. The model anticancer drugs adopted in this work are curcumin and 5-fluorouracil (5-FU). Most of the drug loaded nanofibers synthesized thus far have failed to address the needs of personalized medication due to poor scalability of drug loading and delivery kinetics. This work opens up new avenues for circumventing such complications by altering the drug release profile by a simple one-step crosslinking reaction. With an aim to emphasize the role of polymer crosslinking in drug release kinetics, two variations of dual drug loaded core–shell nanofibers were synthesized with different extents of crosslinking and polymer composition. These two variations of drug loaded nanofibers exhibited contrasting 5-FU release profiles and thus manifested different therapeutic efficacy at different time points against A549 (Non-Small Cell Lung cancer) cells. The drug release profile of these fibers was further corroborated by different kinetic models to gain a perspective on the underlying mechanism driving the drug release from type I and type II nanofibers. The synergistic therapeutic potential of curcumin and 5-FU loaded core–shell nanofibers (type I and type II nanofibers) was also validated against A549 cells. As an outcome of this work, a clear correlation of cell viability with time lag in drug delivery in the case of type I and type II nanofibers could be drawn, which makes nanofiber based drug delivery even more flexible and therapeutically effective with minimal side effects.
 Please wait while we load your content...
Please wait while we load your content...

