DOI:
10.1039/C4RA04920A
(Paper)
RSC Adv., 2014,
4, 33672-33679
Preparation and in vitro release of buccal tablets of naringenin-loaded MPEG-PCL nanoparticles
Received
24th May 2014
, Accepted 28th July 2014
First published on 28th July 2014
Abstract
Naringenin-loaded monomethoxy poly(ethylene glycol)-poly (ε-caprolactone) (MPEG-PCL) nanoparticles (NARNP) and their formulation into buccal tablets using mucoadhesive polymers were developed to improve the solubility of naringenin and to treat oral inflammatory and ulcerative diseases. In our work, physicochemical characterizations of NARNP including particle size, zeta potential, transmission electron microscopy (TEM), differential scanning calorimetery, Fourier transform infrared spectroscopy and in vitro release were studied. NARNP buccal tablets and control buccal tablets containing MPEG-PCL copolymers and mannitol respectively were prepared and they were evaluated by weight variation, hardness, friability and in vitro release. NARNP had a small size (<100 nm), good encapsulation efficiency (95.26 ± 1.15%), and high drug loading (9.95 ± 0.15%). The lyophilized powder of NARNP showed good stability and re-solubility. For the in vitro release study of buccal tablets, Drug dissolution rate was investigated by the USP I method. To simulate in vivo conditions of the oral cavity, a new dissolution test apparatus was designed for assessment of drug release. Compared to control groups, buccal tablets containing NARNP showed more rapid and complete drug release (more than 80%) over 12 h using two dissolution test apparatuses. NARNP incorporated into buccal tablets effectively improved the release of naringenin. With desirable drug release and release time (12 h), NARNP buccal tablets may be an efficient vehicle for treatment of oral inflammatory and ulcerative diseases.
1. Introduction
Naringenin (4′,5,7-trihydroxyflavonone) is a naturally-occurring flavonoid present in citrus fruits1,2 and grapefruit. It has been proved to possess anti-inflammatory,3 anti-atherogenic4 antitumor5 and anticancer effects.6 Previous reports showed naringenin exerts an anti-inflammatory effect by inhibiting nitric oxide and prostaglandin E2 production.7,8 In addition, naringenin exhibited a strong capacity to inhibit pro-inflammatory cytokines,9 such as interleukin-1β, tumor necrosis factor-α and interleukin-6, which contribute to the pathophysiology of oral inflammatory and ulcerative diseases, especially periodontitis.10 Though pharmaceutical applications of naringenin attract great attention, a slow dissolution rate from solid forms restricts its use due to its poor water-solubility. It was reported that the absolute bioavailability of naringenin after oral administration only achieved 4% in rabbits.11 Substantial efforts have been made to improve its water solubility by employing the complex with hydroxypropyl-β-cyclodextrin,12 phospholipid complexes,13 solid dispersions,14 microparticles,15 nanoparticles.16,17
Recently, nanotechnology has been widely applied in drug delivery and accounts for the main part of nanomedicine.18 Polymeric nanoparticles are a promising vehicle with potential to overcome low aqueous solubility of drugs.19 Amphiphilic polymeric molecules can spontaneously form core–shell structures in aqueous medium. Their hydrophobic cores can serve as reservoirs for poorly water-soluble drugs. Encapsulation of hydrophobic drugs into micelles can substantially increase a drug's solubility/dispersibility in aqueous media. Among various amphiphilic copolymers, PEGlation polymers, such as PEGylated polylactide (PLA),20 PEGylated poly(lactide-co-glycolide) (PLGA)21,22 and PEGylated poly(ε-caprolactone) (PCL),23,24 have gained considerable attention for their biodegradability properties, biocompatibility and low toxicity. PEGlation polymers employed as drug carriers through intravascular, oral, nasal, ocular, transdermal administration have been intensively studied.25–29 In the study, to enhance solubility of naringenin, NARNP employing biodegradable amphiphilic copolymers monomethoxy poly(ethylene glycol)-poly(3-caprolactone) (MPEG-PCL) as carriers were prepared.
Buccal mucoadhesive dosage forms have been explored intensively during the last decade due to unique physiological features.30 Buccal route could be used for local delivery as well as systemic delivery. For many oral inflammatory and ulcerative diseases are chronic, it requires chronic treatment approaches. Local drug delivery was considered be to an efficient drug-delivery approach for treatment of oral conditions. These topical formulations could be easily delivered into the site of diseases, reduce potential side effects, improve patients' compliance, exhibit retention within the specific site of action for desired period of time. Several drug candidates31–33 have been studied successfully for topical applications via buccal route. We aim to develop NARNP and their formulation into buccal tablets using mucoadhesive polymers for treatment of oral inflammatory and ulcerative disease.
To the best of our knowledge, no work was reported on naringenin-loaded nanoparticles utilizing MPEG-PCL as carriers. In addition, this was the first time that NARNP compressed into mucoadhesive buccal tablet dosage form were prepared. In present work, we decided firstly to investigate the feasibility of preparing NARNP, using the lyophilized nanoparticles samples to prepare buccal tablets by direct compression method, and finally to investigate the dissolution rate of naringenin. NARNP were prepared by a solvent evaporation method with MPEG-PCL as carriers. Characteristics of NARNP were studied by laser diffraction particle size detector, zeta potential, transmission electron microscope (TEM), differential scanning calorimeter (DSC), FT-IR, in vitro drug release. The buccal tablets were evaluated by weight variation, hardness, friability and in vitro drug release. Drug dissolution rate was investigated by the USP dissolution apparatus. To simulate in vivo conditions of oral cavity, a new dissolution test apparatus was designed for in vitro assessment of buccal tablets with respect to drug release.
2. Materials and methods
2.1 Materials
Naringenin (98% purity) was purchased from Shaanxi Jiahe Phytochem Co., Ltd., China. Milk protein concentrate (MPC85) was gift sample from Beijing he xin xing Tong Trade Company Ltd., China. Hydroxypropyl methylcellulose K4M, corn starch, anhydrous lactose, magnesium stearate, silica powder and mannitol were all obtained from Xi'an Yue Lai Medical Technology Co., Ltd., Shaanxi, China. Acetone was obtained from Tianjin Tianli Chemical Reagent Co., Ltd., Tianjin, China. MPEG-PCL diblock copolymer with a designed molecular weight of 2 k–2 k was obtained from our laboratory. All other chemicals used were analytical or high-performance liquid chromatography (HPLC) grade.
2.2 Preparation of NARNP
NARNP were prepared by a solvent evaporation method.34 To obtain drug-loaded nanoparticles, 150 mg of naringenin and 1.5 g of MPEG-PCL diblock copolymer were co-dissolved in 150 mL acetone. The acetone was removed at 55 °C under reduced pressure on a rotary evaporator (RE-52A, Shanghai Ya Rong Biochemistry Instrument Factory, China) resulting in the formation of a homogenous film. The naringenin-MPEG-PCL film was rehydrated with 45 mL of deionized water at 55 °C with gentle agitation resulting in a clear solution of drug-loaded MPEG-PCL nanoparticles (Fig. 1). The solution was filtered using a 0.22 μm filter. Finally, 40 mL prepared NARNP were lyophilized and stored at 4 °C until use.
 |
| | Fig. 1 Spontaneous naringenin-loaded nanoparticles formation from amphiphilic molecules in aqueous media. | |
2.3 Characterization
2.3.1 Size and zeta potential. Particle size distribution spectra of nanoparticles (blank nanoparticles, NARNP and their re-dissolved nanoparticles) were determined by laser diffraction particle size detector (Nano-ZS, Malvern Instrument, UK). Zeta potential was measured by Malvern Zeta analyzer (Nano-ZS, Malvern Instrument, UK). All measurements were performed at 25 °C. The measurements were performed in triplicate.
2.3.2 Morphology. Morphological examination of nanoparticles was conducted using transmission electron microscope (H–6009IV, Hitachi, Japan). One drop of nanoparticles suspension was negatively stained with phosphotungstic acid, placed on a copper grid covered with nitrocellulose membrane and air-dried before observation.
2.3.3 Drug encapsulation and loading capacity. Drug loading (DL) and encapsulation efficiency (EE) were determined as follows. Briefly, 1 mL of NARNP was freeze-dried to constant weight. Then the lyophilized nanoparticles sample was dissolved in 0.5 mL acetone. The amount of naringenin in the solution was determined by HPLC. Lastly, drug loading (DL) and encapsulation efficiency (EE) of drug loaded nanoparticles were calculated according to eqn (1) and (2):| |
 | (1) |
| |
 | (2) |
2.3.4 FT-IR. Fourier transform infrared (FT-IR) spectra for various samples (free naringenin, MPEG-PCL and NARNP) were obtained on a FT-IR Spectrometer (FTIR-8400S Fourier Transform Infrared Spectrophotometer, Shimadzu, Japan). Samples were mixed with dry crystalline KBr and pellets were prepared. A spectrum was collected for each sample within the wave number region 400–4000 cm−1.
2.3.5 DSC. Free naringenin, MPEG-PCL, naringenin and MPEG-PCL physical mixture and NARNP were recorded using a differential scanning calorimeter (DSC822e, METTLER TOLEDO, Switzerland). For each measurement, an appropriate amount of samples (5–10 mg) was sealed in aluminum pans and scanned in a temperature range of 25 °C to 290 °C with a heating rate of 10 °C min−1.
2.3.6 In vitro release. Free naringenin, NARNP and re-dissolved nanoparticles solution samples were placed in a dialysis bag (molecular weight cutoff, 1 kDa) containing 65 mL of media, and incubated in a water-bath at 37 °C with gentle shaking (100 rpm). At predetermined time points, 0.5 mL aliquots of the solutions were withdrawn from release media. To maintain a constant volume, an equal volume of the same dissolution medium was added back. Afterward, the concentration of naringenin released was monitored by HPLC (XTerra®RP-18 Column, 150 mm × 4.6 mm–5 μm, Waters, America, the mobile phase consisted of methanol–water (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with 0.5% acetic acid, flow rate was 1 mL min−1, the column temperature was 25 °C, the injection volume was 20 μL, and the detector was fixed at a wavelength of 288 nm.). The media for release studies were phosphate-buffered solutions of different pH values (pH 5.5, 6.8 and 7.4) containing Tween-80 (0.5% w/w). This study was repeated three times.
:1) with 0.5% acetic acid, flow rate was 1 mL min−1, the column temperature was 25 °C, the injection volume was 20 μL, and the detector was fixed at a wavelength of 288 nm.). The media for release studies were phosphate-buffered solutions of different pH values (pH 5.5, 6.8 and 7.4) containing Tween-80 (0.5% w/w). This study was repeated three times.
2.4 Formulation of buccal tablets
Tablets were prepared by direct compression technique using HPMC K4M and MPC85 as mucoadhesive polymers. The lyophilized NARNP and excipients except lubricant (magnesium stearate) were mixed in the order of ascending weights and blended. After uniform mixing of ingredients, magnesium stearate was added and mixed again. Tablets (T3, 8 mm diameter and 120 mg weight) were prepared using a single punch tablet press (DP-30, Beijing Gylongli Sci. & Tech. Co., Ltd., China) at 1 ton pressure. To discuss improvement of dissolution rate, control tablets containing naringenin and MPEG-PCL (T2) naringenin and mannitol (T1) were also prepared. In the case of preparation of different buccal tablets, all the parameters during compression are consistent. Compositions of different tablets are listed in Table 1.
Table 1 Composition of different buccal tablets
| Form |
Composition (mg) |
| T1 |
T2 |
T3 |
| MPC85 |
27.43 |
27.43 |
27.43 |
| HPMC K4M |
20.50 |
20.50 |
20.50 |
| Corn starch |
9.20 |
9.20 |
9.20 |
| Anhydrous lactose |
5.57 |
5.57 |
5.57 |
| Magnesium stearate |
1.32 |
1.32 |
1.32 |
| Silica powder |
0.98 |
0.98 |
0.98 |
| Naringenin |
5 |
5 |
— |
| MPEG-PCL |
— |
50 |
— |
| NARNP |
— |
— |
55 |
| Mannitol |
50 |
— |
— |
2.5 Design of the dissolution rate test apparatus
To simulate in vivo conditions of oral cavity, the dissolution rate test apparatus (Fig. 2) was deigned to assess drug release. The dissolution testing device comprises a dissolution cell having a cap functioning to attach a buccal tablet and in which can contain 1.5 mL media, a peristaltic pump which can pump dissolution media from a dissolution media reservoir at about 0.6 mL min−1, a thermostat bath to hold the dissolution cell maintaining at 37 °C, magnetic stirrer located beneath the thermostat bath to control the rotation action of the magnetic needle set in the cell at 50 rpm. We have obtained a patent for this apparatus (China Patent no. 201320445613.7).
 |
| | Fig. 2 Structure of the dissolution rate test apparatus. (1) Dissolution cell, (2) Cap, (3) temperature regulator, (4) thermostat bath, (5) buccal tablet, (6) magnetics stirrer and (7) magnetic needle. | |
2.6 Evaluation of buccal tablets
2.6.1 Weight variation. Twenty tablets were weighed using an electronic balance (AUY220, Shimadzu) and the average weight was calculated.
2.6.2 Hardness. Hardness of the tablets was determined using hardness tester (YD-1, shanghai Develop Machinery Co., Limited). It is expressed in kg. Six tablets were randomly picked from each formulation. The mean and standard deviation values were calculated.
2.6.3 Friability. Friability of the tablets was determined by using friabilator (CJY-300B, shanghai hongyi). After any loose dust were removed, pre-weighed 20 tablets were placed in the friabilator and rotated at 25 rpm for 4 min. Tablets were de dusted using a soft brush and reweighed. The percentage loss was determined.
2.6.4 In vitro drug release studies.
2.6.4.1 Using the USP dissolution apparatus. In vitro drug release studies of mucoadhesive tablets were also carried out in USP dissolution test apparatus, employing 500 mL of artificial saliva ((pH 6.89, 5 mmol dm−3 sodium bicarbonate, 7.36 mmol dm−3 sodium chloride, 20 mmol dm−3 potassium chloride, 6.6 mmol dm−3 sodium dihydrogen phosphate monohydrate and 1.5 mmol dm−3 calcium chloride dehydrate)) as dissolution medium at 37 ± 0.2 °C and 50 rpm maintaining sink conditions. At appropriate time intervals (0.5, 1, 2, 3, 4, 6, 8, 10 and 12 h), 1 mL of samples was withdrawn and an equal volume of medium was added to maintain the volume constant. The samples were filtered and analyzed by HPLC. Six dissolution assays were performed for each formulation.
2.6.4.2 Using the dissolution rate test apparatus. One single tablet was attached to the cap by using 20 μL of artificial saliva and then exerting a press for 30 s. Naringenin released from the buccal tablet was determined by introducing the tablet attached to the cap in the dissolution cell. Artificial saliva was pumped through the cell at about 0.6 mL min−1 using a peristaltic pump and uniform mixing of the 1.5 mL artificial saliva in the cell was provided by magnetic stirrer at 50 rpm. The whole cell was maintained at 37 °C with the help of a thermostat bath and temperature regulator. Fractional samples were collected at 0.5, 1, 2, 3, 4, 6, 8, 10 and 12 h in the collector. After ultrasonic mixing, samples were filtered through a 0.45 μm filter and analyzed by HPLC. Six tablets of each formulation were investigated.
3. Results and discussion
In the present study, we prepared NARNP by a solvent evaporation method, and freeze-dried NARNP into nanoparticles powder for preparation of buccal tablets. As shown in Fig. 3A, NARNP (c) and re-dissolved NARNP solution (e) were transparent similarly to blank nanoparticles (b) obtained by dissolving MPEG-PCL in pure water. These results indicate naringenin was successfully encapsulated into MPEG-PCL nanoparticles and the white lyophilized powder of NARNP (d) with good re-solubility was stable. Poorly water-soluble naringenin was easily encapsulated into polymeric nanoparticles due to hydrophobic interaction .The encapsulation efficiency and drug loading were 95.26 ± 1.15% and 9.95 ± 0.15%, respectively.
 |
| | Fig. 3 (A) Appearance characteristic of nanoparticles. (a) Pure water; (b) blank nanoparticles; (c) NARNP; (d) the lyophilized powder of naringenin-loaded MPEG-PCL nanoparticles; (e) re-dissolved NARNP solution. (B) Different buccal tablets, T1 (buccal tablets prepared with naringenin and mannitol), T2 (buccal tablets prepared with naringenin and MPEG-PCL), T3 (NARNP buccal tablets). | |
3.1 Characterization
3.1.1 Size and zeta potential and morphology. The mean particle size and size distribution of prepared polymeric nanoparticles were determined by DLS. Zeta potential of NARNP was about −9.62 ± 0.58 mv. As shown in Fig. 4A, The size of blank nanoparticles and NARNP were less than 100 nm in diameter with a very narrow particle size distribution. The size of freshly prepared NARNP and re-dissolved NARNP obtained from DLS experiment were 53.86 ± 0.64 nm (PDI = 0.15 ± 0.002), 54.60 ± 0.64 nm (PDI = 0.15 ± 0.002) respectively. The particle size distribution spectrum of freshly blank nanoparticles and re-dissolved nanoparticles were presented, indicating these were monodisperse with a mean diameter of about 70 nm. These results clearly suggest size of NARNP was smaller than blank nanoparticles. We concluded interaction between naringenin and MPEG-PCL 2 k–2 k were considered to affect particle size. The naringenin containing hydroxyl groups may form hydrogen-bonds with the carbonyl groups of PCL. The H-bonding interactions might narrow the size of NARNP.
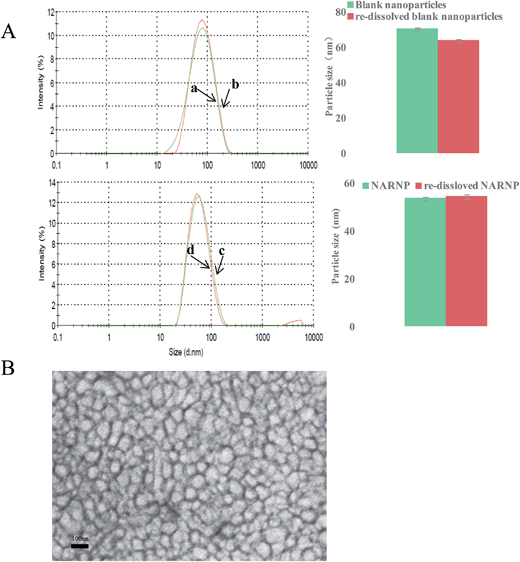 |
| | Fig. 4 (A) The particle size distribution spectrum and the mean particle size of nanoparticles. (a) Blank nanoparticles, (b) re-dissolved blank nanoparticles, (c) NARNP, (d) re-dissolved NARNP. (B) Morphology of NARNP determined by TEM. | |
Fig.4B presents the TEM pictures of nanoparticles. It could be observed that most of nanoparticles exhibited a spherical shape and the size was less than 100 nm in diameter, which was in coincidence with the data from DLS.
3.1.2 FT-IR. The FT-IR spectra of free naringenin, MPEG-PCL and NARNP were presented as shown in Fig. 5A. A major band at 3290 cm−1 and 3117 cm−1 were observed in the spectra of naringenin. These peaks were assigned to O–H stretching. The sharp peaks C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching in the spectrum of naringenin was observed at 1626 cm−1. The FT-IR spectrum of MPEG-PCL showed peaks at 1724 cm−1 due to CO stretching and 1107 cm−1 corresponding to C–O–C stretching vibrations, along with peaks at 2945 cm−1 and 2867 cm−1 due to –CH2 stretching vibration. These peaks were also detected in the spectra of NARNP. However, the position of the peaks corresponding to CO stretching in the spectra of NARNP shifted to higher wave numbers (1639 cm−1), compared to those for naringenin. In the meantime, the position of these peaks due to O–H stretching shifted to higher wave numbers. These differences suggested involvement of hydrogen bonding groups between hydroxyl groups of naringenin and the carbonyl groups of PCL. The intermolecular hydrogen bonds might weakened intramolecular hydrogen bonds between 4-CO and 5-OH in naringenin, resulting in these increased wave numbers. From the above results, it is speculated that hydrogen bonding reaction was achieved in NARNP, which was favorable for the incorporation of naringenin.
O stretching in the spectrum of naringenin was observed at 1626 cm−1. The FT-IR spectrum of MPEG-PCL showed peaks at 1724 cm−1 due to CO stretching and 1107 cm−1 corresponding to C–O–C stretching vibrations, along with peaks at 2945 cm−1 and 2867 cm−1 due to –CH2 stretching vibration. These peaks were also detected in the spectra of NARNP. However, the position of the peaks corresponding to CO stretching in the spectra of NARNP shifted to higher wave numbers (1639 cm−1), compared to those for naringenin. In the meantime, the position of these peaks due to O–H stretching shifted to higher wave numbers. These differences suggested involvement of hydrogen bonding groups between hydroxyl groups of naringenin and the carbonyl groups of PCL. The intermolecular hydrogen bonds might weakened intramolecular hydrogen bonds between 4-CO and 5-OH in naringenin, resulting in these increased wave numbers. From the above results, it is speculated that hydrogen bonding reaction was achieved in NARNP, which was favorable for the incorporation of naringenin.
 |
| | Fig. 5 (A) FT-IR spectra for free naringenin (a), MPEG-PCL (b) and NARNP (c). (B) DSC analysis of naringenin (a), MPEG-PCL (b), naringenin and MPEG-PCL physical mixture (c) and NARNP (d). | |
3.1.3 DSC. DSC analysis is an important approach to determine the state of incorporated drug.35 The DSC curves of naringenin, MPEG-PCL, naringenin and MPEG-PCL physical mixture, and NARNP were shown in Fig. 5B. The DSC scan of naringenin showed an endothermic peak at its melting temperature (approximately 254 °C), MPEG-PCL showed a broad transition at 55 °C. The physical mixture of naringenin and MPEG-PCL exhibited the same exothermic peak as that of naringenin. Furthermore, the DSC profile of NARNP did not exhibit a sharp endothermic peak at 254 °C. The DSC study reveals that encapsulated naringenin are not in an amorphous or disordered crystalline phase of a molecular dispersion or a solid solution state in the polymer matrix as melting peak is absent in the thermograms of NARNP.
3.1.4 In vitro release. As is well known, pH in mouth is around 5.5–7.0 due to saliva with weak buffering capacity. In present study, release of NARNP and free naringenin in different pH values (pH 5.5, 6.8 and 7.4) were investigated. Fig. 6 shows release of free naringenin, naringenin release from NARNP and re-dissolved nanoparticles at different pH values (pH 5.5, 6.8 and 7.4). At pH 5.5, release of free naringenin was more than 95% within 6 h, while at pH 6.8 and 7.4, more than 99% of naringenin was rapidly released within 40 min, 1 h respectively. In the profile of naringenin release from NARNP and re-dissolved nanoparticles, no significant burst release was observed in all of nanoparticles. At pH 6.8 and 7.4, naringenin release from NARNP was more than 85% over 24 h, slower release was observed at pH 5.5 with only 74% of total naringenin released at the end of the study. Naringenin release from re-dissolved nanoparticles showed a dissolution profile mostly similar to that of NARNP at different pH values. Unlike the rapid release of free naringenin, NARNP with good encapsulation showed slow release character. In vitro drug release, the release of naringenin at pH 5.5 was obviously slower than that at pH 6.8 and 7.4. It was found that the hydrolytic degradation of PCL-PEG-PCL caused by cleavage of ester bonds made it unstable in aqueous solution.36,37 The different release at different pH of nanoparticles probably is caused by combined effects of naringenin's acidic nature and degradation of MPEG-PCL at different pH values.
 |
| | Fig. 6 Release profile of free naringenin, naringenin from NARNP and re-dissolved nanoparticles at different pH values. | |
3.2 Evaluation of buccal tablets
As shown in Table 2, the weight variation of tablets was 119 to 121 mg. Weight variation test revealed that the tablets were within the range of Chinese pharmacopoeial limit. Hardness test indicated good mechanical strength. The hardness of prepared buccal tablets was found to be in the range of 2.89 to 3.30 kg. Friability was less than 1% indicated that buccal tablets had a good mechanical resistance.
Table 2 Physicochemical properties of buccal tablets
| FC |
Weight variationa (mg) |
Hardnessa (kg) |
Friabilitya (%) |
| Mean ± SD, n = 3. |
| T1 |
119.53 ± 0.96 |
2.89 ± 0.23 |
0.70 ± 0.21 |
| T2 |
120.86 ± 1.45 |
3.30 ± 0.38 |
0.66 ± 0.16 |
| T3 |
119.56 ± 0.83 |
3.15 ± 0.41 |
0.69 ± 0.09 |
3.3 In vitro drug release studies of buccal tablets
Naringenin, a flavonoid with very slightly water solubility,38 shows incomplete release in simulated biological fluids from oral dosage forms. To increase the solubility, literature reported some studies on naringenin-loaded nanoparticles.39,40 The Eudragit® E (EE) cationic copolymer was mainly employed as nanocarrier. Eudragit E possessing a basic site of dimethylamino group could easily dissolve in aqueous media up to approximately pH 5, which was suitable for drug release in gastric juice.41 However, for nanoparticles buccal tablets delivered into oral cavity, drug was required for ideal release from nanocarrier in neutral environment. In our study, MPEG-PCL was employed as carriers to encapsulate naringenin. We supposed that appropriate drug release could be obtained by preparing buccal tablets with NARNP. As shown in Fig. 3B, NARNP buccal tablets and control tablets were formulated under the same formulation parameters and their dissolution performance were compared. The release profiles of naringenin from bioadhesive tablets using USP dissolution apparatus is shown in Fig. 7A The release rate of drug from formulation prepared with nanoparticles (T3) was extremely significant in comparison to control buccal tablets (T1, T2). The buccal tablets prepared with MPEG-PCL (T2) showed slightly faster release (58.54%) compared to T1 (44.84%) at the end of 12 h. Similar drug release phenomenon of buccal tablets investigated by the dissolution rate test apparatus was found (Fig. 7B). In T3 containing NARNP 84.40% of drug was released at the end of 12 h. The release rate of the nanoparticles buccal tablet was much faster than that T1 and T2 (58.67% and 44.12%, respectively). Naringenin release from T1 was inhibited due to its poor solubility in aqueous media. Compared to T1, sight improvement in dissolution rate of T2 can be attributed to the presence of self-assembling MPEG-PCL copolymers in buccal tablets. At a certain extent, amphiphilic MPEG-PCL copolymers can slightly increase the solubility of drug.
 |
| | Fig. 7 The release profiles of naringenin from bioadhesive tablets different dissolution apparatus. (A) USP dissolution apparatus. (B) The dissolution rate test apparatus. | |
As expected, buccal tablets containing NARNP offered more rapid and complete drug release, this result is attributed to existing forms of naringenin in NARNP. MPEG-PCL may be used as polymeric carrier capable of giving relatively good encapsulation efficiency for poorly soluble drugs.42 With no crystallinity of naringenin encapsulated in the polymer matrix, NARNP significantly increasing dissolution and diffusion of naringenin to the outer solution allowed a faster release in the medium. Buccal tablets employed HPMC and MPC85 with moderate extended release properties as mucoadhesive polymers. From buccal tablets naringenin was slowly released over a period of 12 h. The sustained-release at the site of disease allows to decreasing potential side effects and improving patients' compliance. This buccal tablet is desirable for treatment of oral inflammatory and ulcerative disease.
4. Conclusions
Biodegradable copolymers of MPEG-PCL nanoparticles loaded with naringenin were prepared by a solvent evaporation method. Studies of NARNP showed good characteristics such as a small size, narrow size distribution, good encapsulation efficiency, high drug loading. In addition, it was found that naringenin release was different in the phosphate-buffered solutions of different pH values. It was the first time that we successfully prepared NARNP buccal tablets using lyophilized powder of NARNP with good stability and re-solubility. Good results were obtained in physicochemical characteristics and in vitro release studies. Compared to control tablets, naringenin released from NARNP buccal tablets showed more rapid and complete release character, indicating encapsulation of naringenin into nanoparticles could improve dissolution in the medium. In vitro release study, the desirable release time (12 h) with more than 80% release for buccal tablets made naringenin possible being the delivery of anti-inflammatory medications for treatment of oral conditions.
Acknowledgements
This work was supported by National Natural Science Foundation of China (no. 81072643 and no. 81270347), Natural Science Foundation of Shaanxi Province (2012JQ4025) and Fundamental Research Funds for the Central Universities (XJJ2013054, XJ08142011, XJ08142016 and XJJ2011091).
References
- S. Kawaii, Y. Tomono, E. Katase, K. Ogawa and M. Yano, J. Agric. Food Chem., 1999, 47, 3565–3571 CrossRef CAS PubMed.
- Y. Nogata, K. Sakamoto, H. Shiratsuchi, T. Ishii, M. YANO and H. Ohta, Biosci., Biotechnol., Biochem., 2006, 70, 178–192 CrossRef CAS PubMed.
- M. Hämäläinen, R. Nieminen, P. Vuorela, M. Heinonen and E. Moilanen, Mediators Inflammation, 2007, 2007 Search PubMed.
- C.-H. Lee, T.-S. Jeong, Y.-K. Choi, B.-H. Hyun, G.-T. Oh, E.-H. Kim, J.-R. Kim, J.-I. Han and S.-H. Bok, Biochem. Biophys. Res. Commun., 2001, 284, 681–688 CrossRef CAS PubMed.
- S.-i. Kanno, A. Tomizawa, T. Hiura, Y. Osanai, A. Shouji, M. Ujibe, T. Ohtake, K. Kimura and M. Ishikawa, Biol. Pharm. Bull., 2005, 28, 527–530 CAS.
- D. Arul and P. Subramanian, Biochem. Biophys. Res. Commun., 2013, 434, 203–209 CrossRef CAS PubMed.
- C.-L. Chao, C.-S. Weng, N.-C. Chang, J.-S. Lin, S.-T. Kao and F.-M. Ho, Nutr. Res., 2010, 30, 858–864 CrossRef CAS PubMed.
- K. Shiratori, K. Ohgami, I. Ilieva, X.-H. Jin, K. Yoshida, S. Kase and S. Ohno, J. Ocul. Pharmacol. Ther., 2005, 21, 298–304 CrossRef CAS PubMed.
- H. M. T. Herath, Y. Takano-Ishikawa and K. Yamaki, J. Med. Food, 2003, 6, 365–370 CrossRef PubMed.
- C. Bodet, V. La, F. Epifano and D. Grenier, J. Periodontal Res., 2008, 43, 400–407 CrossRef CAS PubMed.
- S.-L. Hsiu, T.-Y. Huang, Y.-C. Hou, D.-H. Chin and P.-D. L. Chao, Life Sci., 2002, 70, 1481–1489 CrossRef CAS.
- J. Wen, B. Liu, E. Yuan, Y. Ma and Y. Zhu, Molecules, 2010, 15, 4401–4407 CrossRef CAS PubMed.
- A. Semalty, M. Semalty, D. Singh and M. Rawat, J. Inclusion Phenom. Macrocyclic Chem., 2010, 67, 253–260 CrossRef CAS.
- F. Kanaze, E. Kokkalou, I. Niopas, M. Georgarakis, A. Stergiou and D. Bikiaris, J. Therm. Anal. Calorim., 2006, 83, 283–290 CrossRef CAS.
- F. Sansone, P. Picerno, T. Mencherini, F. Villecco, A. D'Ursi, R. Aquino and M. Lauro, J. Food Eng., 2011, 103, 188–196 CrossRef CAS PubMed.
- N. Krishnakumar, N. Sulfikkarali, S. Manoharan and R. M. Nirmal, Mol. Cell. Biochem., 2013, 382, 27–36 CrossRef CAS PubMed.
- F.-L. Yen, T.-H. Wu, L.-T. Lin, T.-M. Cham and C.-C. Lin, Pharm. Res., 2009, 26, 893–902 CrossRef CAS PubMed.
- V. Wagner, A. Dullaart, A.-K. Bock and A. Zweck, Nat. Biotechnol., 2006, 24, 1211–1218 CrossRef CAS PubMed.
- G. Gaucher, P. Satturwar, M.-C. Jones, A. Furtos and J.-C. Leroux, Eur. J. Pharm. Biopharm., 2010, 76, 147–158 CrossRef CAS PubMed.
- E. Blanco, E. A. Bey, Y. Dong, B. D. Weinberg, D. M. Sutton, D. A. Boothman and J. Gao, J. Controlled Release, 2007, 122, 365–374 CrossRef CAS PubMed.
- F. Danhier, N. Lecouturier, B. Vroman, C. Jerome, J. Marchand-Brynaert, O. Feron and V. Preat, J. Controlled Release, 2009, 133, 11–17 CrossRef CAS PubMed.
- M. Garinot, V. Fievez, V. Pourcelle, F. Stoffelbach, A. des Rieux, L. Plapied, I. Theate, H. Freichels, C. Jerome, J. Marchand-Brynaert, Y. J. Schneider and V. Preat, J. Controlled Release, 2007, 120, 195–204 CrossRef CAS PubMed.
- M. Gou, X. Zheng, K. Men, J. Zhang, B. Wang, L. Lv, X. Wang, Y. Zhao, F. Luo, L. Chen, X. Zhao, Y. Wei and Z. Qian, Pharm. Res., 2009, 26, 2164–2173 CrossRef CAS PubMed.
- R. Li, X. Li, L. Xie, D. Ding, Y. Hu, X. Qian, L. Yu, Y. Ding, X. Jiang and B. Liu, Int. J. Pharm., 2009, 379, 158–166 CrossRef CAS PubMed.
- X. Gao, W. Tao, W. Lu, Q. Zhang, Y. Zhang, X. Jiang and S. Fu, Biomaterials, 2006, 27, 3482–3490 CrossRef CAS PubMed.
- G. Gaucher, R. H. Marchessault and J.-C. Leroux, J. Controlled Release, 2010, 143, 2–12 CrossRef CAS PubMed.
- X. Li, Z. Zhang, J. Li, S. Sun, Y. Weng and H. Chen, Nanoscale, 2012, 4, 4667–4673 RSC.
- L. Plapied, N. Duhem, A. des Rieux and V. Préat, Curr. Opin. Colloid Interface Sci., 2011, 16, 228–237 CrossRef CAS PubMed.
- B. Xue, Y. Wang, X. Tang, P. Xie, Y. Wang, F. Luo, C. Wu and Z. Qian, J. Biomed. Nanotechnol., 2012, 8, 80–89 CrossRef CAS PubMed.
- V. F. Patel, F. Liu and M. B. Brown, J. Controlled Release, 2011, 153, 106–116 CrossRef CAS PubMed.
- F. Cilurzo, C. G. Gennari, F. Selmin, J. B. Epstein, G. M. Gaeta, G. Colella and P. Minghetti, Eur. J. Pharm. Biopharm., 2010, 76, 437–442 CrossRef CAS PubMed.
- D. S. Jones, A. D. Woolfson, A. F. Brown, W. A. Coulter, C. McClelland and C. R. Irwin, J. Controlled Release, 2000, 67, 357–368 CrossRef CAS.
- S. Mohamed, S. Muzzammil and K. Pramod, Acta Pharmacol. Sin., 2011, 46, 460–465 CAS.
- R. D. Dabholkar, R. M. Sawant, D. A. Mongayt, P. V. Devarajan and V. P. Torchilin, Int. J. Pharm., 2006, 315, 148–157 CrossRef CAS PubMed.
- Y. Hu, J. Xie, Y. W. Tong and C.-H. Wang, J. Controlled Release, 2007, 118, 7–17 CrossRef CAS PubMed.
- Y. Hu, L. Zhang, Y. Cao, H. Ge, X. Jiang and C. Yang, Biomacromolecules, 2004, 5, 1756–1762 CrossRef CAS PubMed.
- C. Shen, S. Guo and C. Lu, Polym. Adv. Technol., 2008, 19, 66–72 CrossRef CAS PubMed.
- P. Zhang, R. Lin, G. Yang, J. Zhang, L. Zhou and T. Liu, J. Chem. Eng. Data, 2013, 58, 2402–2404 CrossRef CAS.
- N. Krishnakumar, N. K. Sulfikkarali, S. Manoharan and P. Venkatachalam, Spectrochim. Acta, Part A, 2013, 115, 648–653 CrossRef CAS PubMed.
- N. Sulfikkarali, N. Krishnakumar, S. Manoharan and R. M. Nirmal, Pathol. Oncol. Res., 2013, 19, 287–296 CrossRef CAS PubMed.
- M. V. Ramirez-Rigo, M. E. Olivera, M. Rubio and R. H. Manzo, Eur. J. Pharm. Sci., 2014, 55, 1–11 CrossRef CAS PubMed.
- M. Gou, K. Men, H. Shi, M. Xiang, J. Zhang, J. Song, J. Long, Y. Wan, F. Luo and X. Zhao, Nanoscale, 2011, 3, 1558–1567 RSC.
Footnote |
| † Ke Wang and Tingting Liu contributed equally to the study. |
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 









