
Rational design of mimetic peptides based on aldo-ketoreductase enzyme as asymmetric organocatalysts in aldol reactions†
Saadi Bayatab,
Bimo A. Tejoc,
Emilia Abdulmalekab,
Abu Bakar Salleha,
Yahaya M. Normia and
Mohd Basyaruddin Abdul Rahman*ab
aEnzyme and Microbial Technology Research Centre, Universiti Putra Malaysia, 43400 Serdang, Selangor Darul Ehsan, Malaysia. E-mail: basya@upm.edu.my; Tel: +60 38946 6798 Tel: +60 38943 5380
bDepartment of Chemistry, Faculty of Science, Universiti Putra Malaysia, 43400 Serdang, Selangor Darul Ehsan, Malaysia
cCenter for Infectious Diseases Research Surya University, Jl. Scientia, Boulevard Blok U/7, Gading Serpong, Tangerang 15810, Banten, Indonesia
First published on 4th August 2014
Abstract
Peptides as a kind of important chiral scaffold are broadly identified for their obvious advantages, diverse structures and accessibility. Based on promiscuous aldo-keto-reductase enzymes, several mimetic peptides were designed which were synthesized and tested as multifunctional organocatalysts in direct asymmetric aldol reactions. The corresponding aldol products were produced with high yields (up to 97%) and excellent diastereoselectivities (up to 99/1) and enantioselectivities (>98%) under mild reaction selectivity and enantioselectivity. The secondary structures of peptide catalysts provide an understanding of their mechanism.
1. Introduction
The aldol reaction is a fundamental organic reaction in the construction of the C–C bond.1–3 Although, for a couple of decades, enzymes as practical catalysts have been increasingly utilized for organic synthesis due to their simple processing requirements, high selectivity and mild reaction conditions, they have always suffered from the problem of limitation in a broad substrate scope. Type I aldolases, which are natural enzymes, employ the primary amine of a lysine residue under aqueous conditions for enamine formation and aldol reactions.4 Despite the emerging promiscuous enzymes for catalyzing different chemical transformations of natural or non-natural substrates, their low activity in terms of both yield and stereoselectivity impelled the chemists to design small molecules inspired from enzymes.5List et al. demonstrated that L-proline itself could catalyze a direct intermolecular asymmetric aldol reaction with an enamine intermediate.6,7 The use of peptides as organocatalysts significantly increased for various reasons:8–15 they emulate the action of natural enzymes;16,17 their building blocks with inherent chirality and functional diversity are easily available; they are very easy to synthesize via solid phase methodology; finally, they provide a high degree of stability. Some of the derivatives, used as catalysts in aldol reactions, are dipeptides,18,19 which have terminal end primary amino acids or secondary amine like proline.20–22 In addition, Wennemers et al. employed tripeptides using the combinatorial method in asymmetry C–C bond formation with excellent stereoselectivity.23–26 Thus, peptides can be ideal asymmetric organocatalysts due to their diverse structures and functionality and because they are great alternatives to small, rigid organocatalysts and enzymes.27–31 Mimetic peptides and oligopeptides have gained a great deal of attention due to their asymmetric catalytic properties, availability and similarity to natural enzymes.25,32–35 The main challenge in developing asymmetric catalysis lies in how to design the peptides in the mimicking of natural enzymes. Mimetic peptides as asymmetric catalyst should be both efficient and capable of accepting a broad range of substrates. The evidence showed that in the active center of natural aldolases, a primary amino group of a lysine residue that lies in a hydrophobic pocket plays the main role in enamine intermediate construction. In this study, aldo-keto reductase’s (AKRs) active site is very similar to aldolase enzyme.36,37 Therefore, all amino acids which have an important role in the catalytic mechanism of AKRs were determined.
In order to probe the catalytic activity, stereoselectivity of the peptide, the design of a peptide to attain high yield of enantiomer excess was particularly considered. Thus, several mimetic peptides were designed based on the active site of AKRs. To realize such a reaction in a single flask, since water is generally a suitable solvent for enzymatic reactions, the organocatalyst should be activated in water.38 Using water as a reaction medium is another attractive research subject, mainly due to the low cost, safety and the environmentally benign nature of water.39 In the present study, the designing of the best structure based on an enzyme which can be employed in different kinds of asymmetric organic reactions, particularly in C–C bond-forming reactions, was considered. To fulfil this purpose, an aldol reaction, which is one of the most important carbon–carbon bond-forming reactions in organic chemistry, was chosen as a model.
2. Results and discussion
2.1 Design of mimetic peptides based on AKRs
In the field of asymmetric organocatalysis, short peptides and peptide-based molecules have emerged as promising catalysts for a rapidly growing body of reactions. Peptides offer many sites for functional and structural diversities that can be used to generate optimized catalysts. Therefore, peptides may be an ideal compromise between small rigid organocatalysts and enzymes. Designing and synthesizing peptides capable of catalyzing a wide range of substrates asymmetrically, has still remained an immense challenge. The strategy of this study lies in the inspiration of nature’s enzymes. According to similarities of the active site of AKRs to aldolase, a series of peptides were designed and synthesized based on AKRs (PDB = 1VBJ) active site, using a Fmoc-solid phase protocol.The active site of AKRs contains several hydrophilic and hydrophobic amino acid residues which scaffold surround the ligand. As exhibited in Fig. 1, these residues have been distributed in different places and the locations are far from each other. It is very difficult to synthesise this polypeptide by using a manual or automated peptide synthesis method. Therefore, the number of amino acids were reduced to eighteen just by removing some amino acids. In the initial study, three mimetic peptides derived from AKRs, which have subsequent sequences PEAGAIASGVPELFVKLH, PHAGAIASGVPELFVKLH and AGAIASGVPELFVKLH, and called peptides PE16aa, PH16aa and 16aa, respectively, were designed and synthesized. The secondary structure of these peptides was predicted by LOMETS as random and α-helix (Fig. 2).
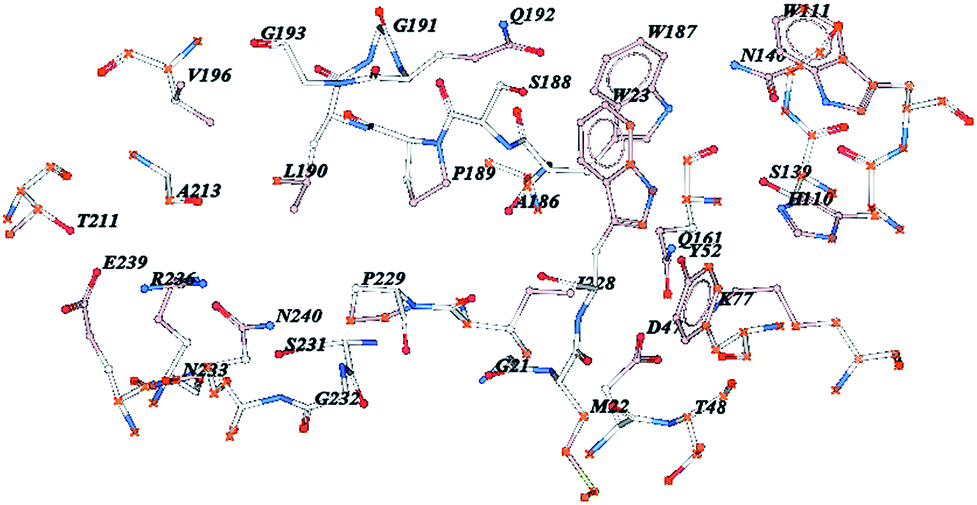 | ||
| Fig. 1 All amino acid residues in AKR’s active site. | ||
 | ||
| Fig. 2 A series of synthesized peptides mimicked from AKRs. | ||
The reaction of p-nitrobenzaldehyde (30 mg, 0.198 mmol, 1 eq.) with cyclohexanone (23.3 mg, 0.238 mmol, 1.2 eq.) was performed as an initial test in the presence of 3 mol% of peptide catalysts in aqueous medium (H2O![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :iPrOH, 1:1, pH = 5.5). The catalytic activity of PH16aa, PE16aa and 16aa was evaluated (Table 1). In order to broaden the range of substrates, the best catalytic efficiency for the defined conditions of the reaction with PH16aa was found (Table 1, entries 9–22). Using PE16aa as an organocatalyst under defined conditions, it was observed that the aldol reaction gave high yield (up to 89%) of the corresponding products, and with moderate enantioselectivity (up to 68%) and diastereoselectivity (anti/syn, up to 99:1) (entries 2–8). These results suggest that the carboxylic side chain of glutamic acid and the imidazole group of histidine can influence enantioselectivity due to hydrogen bonding and the imidazole group is more effective than the carboxylic group.40 Interestingly, when PH16aa was applied as a catalyst in the reaction between aromatic aldehydes and cyclohexanone, the yield and stereoselectivity increased (Table 1, entries 9–17, up to 95% yield, up to 99/1 dr, up to 86% ee). The reaction between p-nitrobenzaldehyde and cyclohexanone was also performed in the presence of 1% sodium dodecyl sulfate (SDS) as an additive, and the results exhibited almost equal yield and %ee as compared to those of the reactions in which SDS was not used. Notably, in most cases, the anti-aldol products were obtained with excellent diastereoselectivity and good enantioselectivity, regardless of the electronic nature of the aromatic aldehydes and ketones. The substitution of aromatic aldehydes and acetone as acyclic ketone catalyzed by PH16aa produced excellent yields and moderate enantioselectivity up to 56% (Table 1, entries 18–22). To investigate the effect of peptide length on diastereo- and enantioselectivity in aldol reactions, a peptide with sixteen amino acids was synthesized, which consisted of a sequence derived from the active site of the AKR enzyme (16aa). This fragment was initiated by a primary amino acid. This polypeptide was designed in order to investigate the roles of the primary amine and side chain in the enhancement of enantioselectivity. The second amino acid was glycine which did not contain carboxylic, imidazol or hydrophilic side function groups for creating strong hydrogen bonds with substrates. The aldol reaction between p-nitrobenzaldehyde and cyclohexanone produced moderate yield and poor enantioselectivity (Table 1, entry 1).
:iPrOH, 1:1, pH = 5.5). The catalytic activity of PH16aa, PE16aa and 16aa was evaluated (Table 1). In order to broaden the range of substrates, the best catalytic efficiency for the defined conditions of the reaction with PH16aa was found (Table 1, entries 9–22). Using PE16aa as an organocatalyst under defined conditions, it was observed that the aldol reaction gave high yield (up to 89%) of the corresponding products, and with moderate enantioselectivity (up to 68%) and diastereoselectivity (anti/syn, up to 99:1) (entries 2–8). These results suggest that the carboxylic side chain of glutamic acid and the imidazole group of histidine can influence enantioselectivity due to hydrogen bonding and the imidazole group is more effective than the carboxylic group.40 Interestingly, when PH16aa was applied as a catalyst in the reaction between aromatic aldehydes and cyclohexanone, the yield and stereoselectivity increased (Table 1, entries 9–17, up to 95% yield, up to 99/1 dr, up to 86% ee). The reaction between p-nitrobenzaldehyde and cyclohexanone was also performed in the presence of 1% sodium dodecyl sulfate (SDS) as an additive, and the results exhibited almost equal yield and %ee as compared to those of the reactions in which SDS was not used. Notably, in most cases, the anti-aldol products were obtained with excellent diastereoselectivity and good enantioselectivity, regardless of the electronic nature of the aromatic aldehydes and ketones. The substitution of aromatic aldehydes and acetone as acyclic ketone catalyzed by PH16aa produced excellent yields and moderate enantioselectivity up to 56% (Table 1, entries 18–22). To investigate the effect of peptide length on diastereo- and enantioselectivity in aldol reactions, a peptide with sixteen amino acids was synthesized, which consisted of a sequence derived from the active site of the AKR enzyme (16aa). This fragment was initiated by a primary amino acid. This polypeptide was designed in order to investigate the roles of the primary amine and side chain in the enhancement of enantioselectivity. The second amino acid was glycine which did not contain carboxylic, imidazol or hydrophilic side function groups for creating strong hydrogen bonds with substrates. The aldol reaction between p-nitrobenzaldehyde and cyclohexanone produced moderate yield and poor enantioselectivity (Table 1, entry 1).
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Cat. | Yieldb (%) | drc (%) | eed (%) |
| a Reaction conditions: aldehydes (30 mg, 0.198 mmol, 1 eq.) with cyclohexanone or acetone (23.3 mg, 0.238 mmol, 1.2 eq. or 1 mL), catalyst (3 mol%), solvent (H2O:iPrOH, 1:1 or H2O:acetone, 1:1), pH = 5.5, RT, 24 h.b Isolated yield.c dr values were determined by the crude product 1H NMR.d ee values were determined by HPLC using a Chiral OD-H and AD-H columns. |
||||||
| 1 | 4-NO2 | Cyclohexanone | 16aa | 67 | 97/3 | 39 |
| 2 | 4-NO2 | Cyclohexanone | PE16aa | 88 | 99/1 | 63 |
| 3 | 2-NO2 | Cyclohexanone | PE16aa | 87 | 98/2 | 61 |
| 4 | 4-Cl | Cyclohexanone | PE16aa | 89 | 96/4 | 62 |
| 5 | 4-CN | Cyclohexanone | PE16aa | 87 | 97/3 | 67 |
| 6 | 4-CF3 | Cyclohexanone | PE16aa | 89 | 99/1 | 65 |
| 7 | 4-Br | Cyclohexanone | PE16aa | 86 | 97/3 | 57 |
| 8 | 2-Cl | Cyclohexanone | PE16aa | 84 | 99/1 | 68 |
| 9 | 4-NO2 | Cyclohexanone | PH16aa | 95 | 95/5 | 86 |
| 10 | 2-NO2 | Cyclohexanone | PH16aa | 94 | 99/1 | 81 |
| 11 | 4-Cl | Cyclohexanone | PH16aa | 92 | 99/1 | 78 |
| 12 | 2-Cl | Cyclohexanone | PH16aa | 95 | 99/1 | 71 |
| 13 | 4-CN | Cyclohexanone | PH16aa | 88 | 99/1 | 81 |
| 14 | 4-CF3 | Cyclohexanone | PH16aa | 93 | 99/1 | 80 |
| 15 | 4-Br | Cyclohexanone | PH16aa | 94 | 99/1 | 84 |
| 16 | H | Cyclohexanone | PH16aa | 92 | 99/1 | 76 |
| 17 | 4-NO2 (Solv. = iPrOH/1% SDS) | Cyclohexanone | PH16aa | 94 | 99/1 | 81 |
| 18 | 4-NO2 | Acetone | PH16aa | 93 | — | 56 |
| 19 | 2-NO2 | Acetone | PH16aa | 92 | — | 45 |
| 20 | 4-Cl | Acetone | PH16aa | 92 | — | 43 |
| 21 | 4-CF3 | Acetone | PH16aa | 94 | — | 39 |
| 22 | 4-Br | Acetone | PH16aa | 94 | — | 35 |
| 23 | 4-NO2 | Cyclohexanone | No cat. | — | — | — |
For further investigation of the peptide length efficacy on an asymmetry aldol reaction, 16aa was cleaved to a shorter peptide to obtain a peptide with a secondary amine at the N-terminus (see the sequences in Fig. 2). The sixteen amino acid polypeptide (16aa) was shortened to eight amino acids (PELFVKLH, 8aa) and was used as a catalyst in the model reaction. The corresponding product catalyzed by this peptide was attained with excellent yield and enantioselectivity (yield = 97%, ee = 97%). This significant result achieved prompted the researchers to investigate the influence of shorter peptides derived from 8aa on the catalytic activity of catalysts in an aldol reaction. Later, five new peptides were designed and synthesized, named 8aa-z, 5aa, 3aa, Fmoc-3aa-R, and 3aa(z)-R (Fig. 2). The theory behind these designs was as following:
8aa-z was designed with the purpose of knowing if the side chain of the primary amine of lysine does have any effect on the enhancement of stereoselectivity. In order to accomplish this aim, the primary amine was protected by benzylchloroformate (Fig. 2, 8aa-z).
Pentapeptide 5aa was designed to investigate the effect of hydrophobic residues such as leucine, phenylalanine, valine, attached to a polar amino acid like glutamic acid, on the catalytic activity, and on the lack of hydrophilic residues such as lysine and histidine. Furthermore, in order to find to what extent the primary amino acid attached to the hydrophobic amino acid followed by a polar amino acid can influence stereoselectivity, the tripeptide 3aa was designed. In Fmoc-3aa-R, the Fmoc group protected the N-terminus of lysine while the primary amine side chain of lysine was free. This tripeptide was attached to a resin. The purpose was to identify the role of free amines on the side chain of lysine in the stereoselectivity of the carbon–carbon forming reaction. Finally, 3aa(z)-R was designed to contain an N-terminus which is free, a carboxybenzyl group which protected the primary amine in the side chain of lysine, and a C-terminus attached to the rink amide-am-resin. The reaction of p-nitrobenzaldehyde with cyclohexanone was conducted using the peptides 8aa, 8aa-z, 5aa, 3aa, Fmoc-3aa-R and 3aa(z)-R as asymmetric catalysts in the defined conditions. Compared to the catalyst 8aa, the yield and enantioselectivity remarkably diminished by almost 10% while the catalyst 8aa-z was utilized as an asymmetry catalyst (Table 2, entry 3). From a mechanistic point of view, this examination showed that primary amine of lysine has an impressive synergy with other amino acid residues to accelerate both the yield and stereoselectivity (Table 2, entry 3). Likewise, the pentapeptide 5aa was evaluated for a direct aldol reaction between p-nitrobenzaldehyde and cyclohexanone in the defined conditions. Although high to excellent yields and stereoselectivities were achieved, its catalytic activity was lower than 8aa (Table 2, entry 4 vs. entry 2). The reason behind this design was that it was envisaged the hydrophobic amino acid residues also play a key role in the enhancement of the yield and stereoselectivity due to steric hindrance. Based on a study by Kofoed et al., Pro-Glu-NH2 and Pro-Asp-NH2 were ineffective both in terms of catalysis and enantioselectivity.16 Surprisingly, when the tripeptide 3aa was employed as a catalyst for a direct aldol reaction between p-nitrobenzaldehyde and cyclohexanone, good yield and stereoselectivity were obtained (Table 2, entry 5) but still lower than 8aa.
| Entry | Cat. | Yield (%) | ee (%) |
|---|---|---|---|
| a Reaction conditions: p-nitrobenzaldehydes (30 mg, 0.198 mmol, 1 eq.) with cyclohexanone (23.3 mg, 0.238 mmol, 1.2 eq.), catalyst (3 mol%), solvent (H2O:iPrOH, 1:1), pH = 5.5, RT. |
|||
| 1 | 16aa | 67 | 39 |
| 2 | 8aa | 97 | 97 |
| 3 | 8aa-z | 89 | 86 |
| 4 | 5aa | 86 | 90 |
| 5 | 3aa | 90 | 84 |
| 6 | Fmoc-3aa-R | 90 | 3 |
| 7 | 3aa(z)-R | 85 | 75 |
On the other hand, although Fmoc-3aa-R gave high yield of the corresponding aldol product, the enantioselectivity dropped to 3% (Table 2, entry 6). This demonstrates that in 3aa, the N-terminus of the lysine formed enamine intermediate properly and the substrate made a hydrogen bonding with histidine. In Fmoc-3aa-R, the distance between the enamine intermediate and histidine is far enough to form a hydrogen-bonding interaction with the imidazole group. Therefore, the corresponding almost racemic aldol product was obtained. The Fmoc protecting group was then removed and the side chain amine group was protected to achieve 3aa(z)-R. High yield and good stereoselectivity were observed (Table 2, entry 7). The flexibility of the free peptide induced higher enantioselectivity, in contrast to the peptide attached to the resin (Table 2, entry 7 vs. 5).
For subsequent reactions, the catalyst 8aa was used to optimize the experimental parameters of the condition, scope, limitations, and solvents. The optimized reaction conditions were screened for enantioselective aldol reactions between cyclohexanone (23.3 mg, 0.238 mmol, 1.2 eq.) and p-nitrobenzaldehyde (30 mg, 0.198 mmol, 1 eq.) in the presence of the octapeptide. TFA salt was chosen as an asymmetric catalyst (3.0 mol%). The iPrOH was applied as a solvent and NMM (N-methylmorpholine) was used to adjust pH = 5.0–5.5. In the presence of just iPrOH, no reaction occurred due to the insolubility of the catalyst. Various additives, such as water, amines, and carboxylic acids, increased the yield and ee of the proline catalyzed aldol reactions.41 The best mole ratio was the mixture of 0.6 mL water with 0.4 mL iPrOH. The reaction proceeded with high yield and enantioselectivity (97%).
The effects of several solvents on the reaction with cyclohexanone and p-nitrobenzaldehyde were also investigated (Table 3). Due to the poor solubility of 8aa in most solvents, reactions mediated by 8aa are limited to polar organic solvents, such as water, dimethyl sulfoxide (DMSO), N,N-dimethylformamide (DMF) and N-methyl-2-pyrrolidone (NMP). No reaction occurred when toluene, chloroform, and THF were used (Table 3, entries 1–3). Additionally, high yield and enantioselectivity were obtained when DMF, DMSO, and NMP were utilized (Table 3, entries 4–6). While DMSO and DMF are commonly used for aldol reactions catalyzed by peptides, these solvents are generally considered problematic for large-scale reactions due to the inconvenient work-up and solvent removal and recovery. The mixture of H2O/DMF (3:1) produced the highest stereoselectivity (Table 3, entry 7). This result indicates that water (as an eco-friendly solvent) is requisite for achieving excellent diastereoselectivity and enantioselectivity. Similar results were achieved with brine (Table 3, entry 8).
| Entry | Solvent | Time (h) | Yield (%) | ee (%) |
|---|---|---|---|---|
| a Reaction conditions: p-nitrobenzaldehydes (30 mg, 0.198 mmol, 1 eq.) with cyclohexanone (23.3 mg, 0.238 mmol, 1.2 eq.), catalyst (3 mol%), solvent, pH = 5.5, RT. | ||||
| 1 | CHCl3 | 72 | NR | — |
| 2 | Toluene | 72 | NR | — |
| 3 | THF | 72 | NR | — |
| 4 | DMF | 24 | 94 | 86 |
| 5 | DMSO | 24 | 95 | 87 |
| 6 | NMP | 24 | 92 | 89 |
| 7 | H2O/DMF (3:1) |
24 | 96 | 97 |
| 8 | Brine/DMF (3:1) |
24 | 91 | 95 |
| 9 | 1% SDS/iPrOH(3:2) |
24 | 93 | 82 |
The model reaction was also performed in SDS (1% w/v)/iPrOH (3:2). As shown in Table 3, entry 9, this mixture is also a reasonable choice for a reaction which will have high yield and good enantioselectivity. The scope and limitations of a direct aldol reaction of cyclohexanone with various aromatic aldehydes, catalyzed by 8aa, were explored (Table 4). Benzaldehydes were substituted with p-nitro, p-trifluoromethyl, p-cyano, p-chloro, o-chloro and o-nitro as electron-withdrawing groups and p-methoxy as an electron-donating group. Electron-withdrawing groups on aromatic ring substrates tend to accelerate the reaction. This is illustrated by the high turnover (3 mol% of catalyst loading) and short reaction time (24 h; Table 4, entries 1–8 and 10). The electron-donating substituent resulted in high enantioselectivity with moderate yield (Table 4, entry 11). The aldol reaction of o-chlorobenzaldehyde and cyclohexanone produced 96:4 dr, >98% ee, and excellent yield (Table 4, entry 4), while benzaldehyde itself reacted with good yield and enantioselectivity (Table 4, entry 9). Pyridinecarboxyaldehyde is a good substrate as well, affording high ee’s (Table 4, entry 8). The aldol reactions of electron-deficient benzaldehydes with cyclohexanone proceeded smoothly to produce aldol adducts with excellent diastereoselectivities (99/1) and enantioselectivities (>98%). It is also interesting to note that both the para- and ortho- substitutions resulted in high yields with good to excellent enantio- and diastereoselectivities.
|
||||||
|---|---|---|---|---|---|---|
| Entry | R1 | R2 | Time (h) | Yieldb (%) | drc (%) | eed (%) |
| a Reaction conditions: p-nitrobenzaldehydes (30 mg, 0.198 mmol, 1 eq.) with cyclohexanone or acetone (23.3 mg, 0.238 mmol, 1.2 eq. or 1 mL), catalyst (3 mol%), solvent (H2O:iPrOH, 1:1 or H2O:acetone, 1:1), pH = 5.5, RT.b Isolated yield.c dr values were determined by crude product 1H NMR.d ee values were determined by HPLC using a Chiral OD-H and AD-H columns. |
||||||
| 1 | 4-NO2 | Cyclohexanone | 24 | 97 | 90/10 | 97 |
| 2 | 2-NO2 | Cyclohexanone | 24 | 94 | 90/10 | 89 |
| 3 | 4-Cl | Cyclohexanone | 24 | 95 | 93/7 | 96 |
| 4 | 2-Cl | Cyclohexanone | 24 | 96 | 96/4 | >98 |
| 5 | 4-CN | Cyclohexanone | 24 | 92 | 99/1 | 86 |
| 6 | 4-CF3 | Cyclohexanone | 24 | 94 | 98/2 | 86 |
| 7 | 4-Br | Cyclohexanone | 24 | 93 | 99/1 | 80 |
| 8 | 4-Pyridinecaboxyaldehyde | Cyclohexanone | 24 | 95 | 99/1 | 98 |
| 9 | H | Cyclohexanone | 24 | 94 | 98/2 | 87 |
| 10 | 4-NO2 | Cycloheptanone | 30 | 71 | 92/8 | 88 |
| 11 | 4-OMe | Cyhexanone | 48 | 65 | 99/1 | 95 |
| 12 | 4-NO2 | Acetone | 15 | 97 | — | 77 |
| 13 | 2-NO2 | Acetone | 15 | 96 | — | 59 |
| 14 | 4-Cl | Acetone | 15 | 96 | — | 52 |
| 15 | 4-CF3 | Acetone | 15 | 96 | — | 74 |
| 16 | 4-Br | Acetone | 15 | 96 | — | 73 |
| 17 | 4-NO2 (no cat.) | Cyclohexanone | 24 | — | — | — |
To examine the generality of the current process, the aldol reactions between aromatic aldehydes and different ketones, including cyclic and acyclic ketones were tested (Table 4, entries 10, 12–16). The diastereo- and enantioselectivities are significantly influenced by the ketone structure. Acyclic ketones produced excellent yields and moderate to good stereoselectivities (Table 4, entries 12–16, up to 77% ee). When acetone was utilized, the enantioselectivity diminished notably, but the reaction was faster and completed in only 15 h. The lower ee% is due to the flexibility and free rotation of acetone. As illustrated by the transition state (Fig. 2), the specific rigid conformation of cyclohexanone results in a remarkably higher enantio- and diastereoselectivity ratio as compared to acyclic ketones. However, the difference in enantioselectivity ratio between p-nitrobenzaldehyde and benzaldehyde in the reaction with cyclohexanone could be due to an electron-withdrawing group; however, it is important to note the key role of strong hydrogen bonding between carboxylic acid and a nitro group, and a carbonyl group with an imidazol group to produce higher ee.42 It seems that the side chain amine group of lysine has a significant role in the transition state to enhance enantioselectivity. When the peptides 8aa(z) and 5aa were employed in the reaction between p-nitrobenzaldehyde and cyclohexanone as catalysts under defined conditions, the enantioselectivity reduced by 10% (Table 4, entries 3 and 4).
The next stage of the investigation was the exploration of the reusability of 8aa as asymmetric catalyst in an aldol reaction. The catalyst can be easily separated through precipitation by adding diethyl ether, ethyl acetate or other low polar solvents. The reusability of the catalyst was evaluated through using cyclohexanone with p-nitrobenzaldehyde. The recovered 8aa could be reused ten times without an obvious loss of enantioselectivity and decreased activity (Table 5, entries 1–10). After the 10th cycle, the activity began to decrease, which could be attributed to the loss of catalyst during recycling.
| Entry | Cat. | Time (h) | Yieldb (%) | eec (%) |
|---|---|---|---|---|
| a Reaction conditions: p-nitrobenzaldehydes (30 mg, 0.198 mmol, 1 eq.) with cyclohexanone or acetone (23.3 mg, 0.238 mmol), catalyst (3 mol%), solvent (H2O:iPrOH, 1:1), pH = 5.5, RT.b Isolated yield.c ee values were determined by HPLC using a Chiral OD-H and AD-H columns. |
||||
| 1 | Run 1 | 24 | 94 | 97 |
| 2 | Run 2 | 24 | 94 | 96 |
| 3 | Run 3 | 24 | 93 | 96 |
| 4 | Run 4 | 24 | 92 | 94 |
| 5 | Run 5 | 24 | 92 | 93 |
| 6 | Run 6 | 24 | 93 | 93 |
| 7 | Run 7 | 24 | 92 | 91 |
| 8 | Run 8 | 24 | 90 | 87 |
| 9 | Run 9 | 24 | 87 | 81 |
| 10 | Run 10 | 24 | 83 | 76 |
3. Mechanism study
These finding suggest a plausible mechanism, similar to that proposed by the groups of List and Houk for proline catalysis, involving enamine formation, a subsequent reaction with the ketone and proton transfer from the carboxylic acid.43,44In conclusion, the multifunctionality properties of 8aa, which is comprised of hydrophilic and hydrophobic amino acid residues, may cause obtaining very high yield and stereoselectivity. The proposed mechanism is presented in Fig. 3.
 | ||
| Fig. 3 Proposed aldol reaction mechanism catalyzed by 8aa. | ||
The secondary structural study of 8aa, showed a random structure. One of the configurations of 8aa is β-turn. Therefore, aldehyde can take place in the pocket of the peptides. Based on experimental data, the primary amine and carboxylic acid of the side chain of lysine and glutamic acid residues had an impressive hydrogen bond interaction with the nitro group of the aldehyde on one side and, on the other side, a hydrogen bond interaction between the imidazole group of histidine and the carbonyl group of the aldehyde helped to keep the substrate in a favoured position. Finally, the nucleophile will attack the carbonyl group on the opposite side to produce a corresponding anti-aldol product.
3.1 Peptide structure studies
Circular dichroism (CD) spectropolarimetry and FT-IR spectroscopy were applied to find the secondary structure of 8aa. The results from computational modelling predicted by LOMETS (LOcal MEta-Threading-Server), indicated a random structure for the catalyst. The CD spectra were performed in two solvents, namely water and 1% SDS. As can be seen in the CD spectra (Fig. 4), a random peptide structure was revealed. The peptide was dissolved in water and the pH was acidic due to the TFA residue. When SDS (1%) was used as a solvent, two negative peaks at approximately 205 and 220 nm were observed, representing a α-helix structure for the peptide, which indicated that the structure of the peptide could change. According to these results, both structures can be catalytically active. The CD data were analyzed by http://perry.freeshell.org/raussens.html, the free online program. The obtained results in water showed that the highest percentage of the peptide’s structure was random (40%), followed by beta sheets (32%) and almost equal amounts of α-helix and β-turn structures (12%). In 1% SDS, the highest percentages of the structures were random followed by α-helix (34 and 31%, respectively) with almost equal amounts of beta sheets (14%) and β-turns (12%) (Fig. 4). | ||
| Fig. 4 CD spectrum of peptide 8aa, in water and 1% SDS. | ||
In addition to CD, the secondary structure of 8aa was investigated via infrared (IR) spectroscopy, as it is also one of the oldest and well-established techniques for the analysis of the secondary structures of polypeptides and proteins.45,46 The amide I band between 1600 and 1700 cm−1 was the most intense absorbance band for all the investigated proteins and peptides, being mainly associated with the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching vibration and directly related to the backbone conformation. This band had a characteristic shape for each peptide investigated. Amides I and II (1500–1600 cm−1) are the two major bands of the protein infrared spectrum and are conformationally sensitive. Amide II results mostly from the N–H bending vibration and from the C–N stretching vibration (18–40%).47,48 Generally, the 1655 cm−1 peak, which is assigned to the α-helix conformation, was positively correlated with bands at 1175, 1305, 2950 and 3330 cm−1, as previously reported.49 The band observed at 1668 cm−1 was assigned to β-turns. Antiparallel β-sheets (1698 cm−1) were not observed. 8aa had an intense absorption peak at 1624–1642 cm−1, corresponding to its high content of β-sheet conformers, which is consistent with amide II band absorptions around 1534 cm−1.50 The 1662 cm−1 signal was assigned to β-turn conformers, which overlapped with a β-sheet. The FT-IR spectra of the octapeptide displayed a large band in the range of 1590 to 1700 cm−1, with maximum absorption at 1648 ± 2 cm−1 (random coil overlap with the β-sheet), and 1668 − 62 cm−1 (β-sheet, β-turn, overlap with α-helix). The 1656 and 1668 cm−1 bands correlated with important amide II bands at 1541, 545 and 1528 cm−1 (Fig. 5). The FT-IR spectroscopy complements CD-spectroscopy according to the conformational studies of peptides and proteins, and its results strongly confirm the CD spectroscopy’s results. The secondary structure of PH16aa in water indicated that the majority was structurally random (39%), followed by the beta sheet (28%) and the amounts of α-helix and β-turn structures, 15 and 12%, respectively (Fig. 6). In 1% SDS, most of the structures were random and α-helix (31% and 38% respectively), with almost equal amounts of beta sheets (12%) and β-turns (12%). The CD spectra for PE16aa and 16aa in both environments (water and SDS) are roughly similar to PH16aa. A CD spectrum for PH16aa is shown in Fig. 6 and those for PE16aa and 16aa are in the ESI.†
O stretching vibration and directly related to the backbone conformation. This band had a characteristic shape for each peptide investigated. Amides I and II (1500–1600 cm−1) are the two major bands of the protein infrared spectrum and are conformationally sensitive. Amide II results mostly from the N–H bending vibration and from the C–N stretching vibration (18–40%).47,48 Generally, the 1655 cm−1 peak, which is assigned to the α-helix conformation, was positively correlated with bands at 1175, 1305, 2950 and 3330 cm−1, as previously reported.49 The band observed at 1668 cm−1 was assigned to β-turns. Antiparallel β-sheets (1698 cm−1) were not observed. 8aa had an intense absorption peak at 1624–1642 cm−1, corresponding to its high content of β-sheet conformers, which is consistent with amide II band absorptions around 1534 cm−1.50 The 1662 cm−1 signal was assigned to β-turn conformers, which overlapped with a β-sheet. The FT-IR spectra of the octapeptide displayed a large band in the range of 1590 to 1700 cm−1, with maximum absorption at 1648 ± 2 cm−1 (random coil overlap with the β-sheet), and 1668 − 62 cm−1 (β-sheet, β-turn, overlap with α-helix). The 1656 and 1668 cm−1 bands correlated with important amide II bands at 1541, 545 and 1528 cm−1 (Fig. 5). The FT-IR spectroscopy complements CD-spectroscopy according to the conformational studies of peptides and proteins, and its results strongly confirm the CD spectroscopy’s results. The secondary structure of PH16aa in water indicated that the majority was structurally random (39%), followed by the beta sheet (28%) and the amounts of α-helix and β-turn structures, 15 and 12%, respectively (Fig. 6). In 1% SDS, most of the structures were random and α-helix (31% and 38% respectively), with almost equal amounts of beta sheets (12%) and β-turns (12%). The CD spectra for PE16aa and 16aa in both environments (water and SDS) are roughly similar to PH16aa. A CD spectrum for PH16aa is shown in Fig. 6 and those for PE16aa and 16aa are in the ESI.†
 | ||
| Fig. 5 Assignment of the FT-IR spectrum of peptide 8aa. | ||
 | ||
| Fig. 6 CD spectrum of peptide PH16aa, in water and 1% SDS. | ||
As can be seen from the FT-IR spectrum related to PH16aa, the band observed at 1654 cm−1 was assigned to an α-helix. Antiparallel β-sheets (1698 cm−1) were not observed. PH16aa had an intense absorption at 1624 cm−1, corresponding to its high content of β-sheet conformers which is consistent with amide II band absorptions around 1536 cm−1. The 1662 cm−1 signal was assigned to β-turn conformers which overlapped with the α-helix (Fig. 7). The FT-IR spectra of PH16aa displayed a large band in the range of 1590 to 1700 cm−1, with some maxima of absorption at 1648 ± 2 cm−1 (a random coil overlap with the β-sheet and α–helix), and 1668 − 62 cm−1 (β-sheet, β-turn, overlap with the α-helix).51 The 1654 and 1668 cm−1 bands correlate with important amide II bands at 1541–1546 cm−1 (Fig. 7). The FT-IR spectra of 16aa and PE16aa confirmed that the structures of these oligopeptides are mostly α-helix and random. The results were almost identical to PH16aa (their spectra can be seen in the ESI†). FT-IR spectroscopy complements CD-spectroscopy according to the conformational studies of peptides and proteins, and its results strongly confirm the CD spectroscopy’s results.
 | ||
| Fig. 7 Secondary structure study of PH16aa using the FT-IR technique. | ||
4. Experimental
4.1 General information
All chemicals were purchased and used without further purification. Analytical thin layer chromatography (TLC) was performed using a Merck 60 F254 precoated silica gel plate (0.2 mm thickness). Flash chromatography was performed using Merck silica gel 60 (70–230 mesh). Fourier transform infrared spectroscopy (FTIR), Perkin Elmer Spectrum 100, was used for identification of the functional groups. NMR data were recorded at 500 MHz for 1H NMR and at 100 MHz for 13C NMR (JEOL JNM ECA) spectrometer. The relative and absolute configurations (dr) of the Aldol reactions were determined through a comparison using 1H NMR spectroscopic analysis. The mass spectra (MS) were measured with a spectrometer (DIMS QP5050A SHIMADZU). The enantioselectivity was determined using HPLC (Waters 1525 Binary Pump and UV-Water 2489) and Chiral OD-H or AD-H columns. The CD spectra were measured by a JASCO J-810 automatic recording spectropolarimeter.4.2 Catalyst preparation
All peptides which were used in this study, were synthesized according to the Fmoc solid-phase strategy.52 The peptide was manually synthesized from an Fmoc-Rink-Amide-Am-Resin, using a 3-fold molar excess of amino acid derivatives with HCTU/DIPEA as a coupling reagent and double coupling cycles. At the end of the synthesis, the peptide was separated from the resin using a mixture of trifluoroacetic acid (TFA, 92.5%), triisopropylsilane (2.5%), 1,4-dithiothreitol (2.5%) and water (2.5%). 10 mL of this solution was utilized with 1 g of resin and agitated for 2 h. The peptide was precipitated in dry diethylether and lyophilized. The purity of the peptides was measured by analytical Waters HPLC (Binary HPLC pump 1525 and UV-Waters 2489), (RPC18, Xbridge 4.6 mm × 250 mm).4.3 CD spectra
The CD spectra were measured on a JASCO J-810 automatic recording spectropolarimeter calibrated with camphorsulfonic acid. The spectra were recorded over 190–240 nm using a 0.1 cm path length quartz cuvette. The scan speed was 100 nm min−1. The curves were digitally recorded and fed through a data processor for signal averaging (3 times) and baseline subtraction. The concentration of the samples was 0.0006 mol L−1 and the CD spectra were run at 20 °C. The program which was retrieved from http://perry.freeshell.org/raussens.html was used for secondary structure analysis.4.4 General procedure for aldol reactions
The corresponding catalysts (3 mol%), NMM (N-methylmorpholine) (1 drop, pH = 5.0–5.5) and iPrOH (0.4 mL) were added to 0.6 mL of water. The reaction mixture was stirred for 20 min followed by addition of the corresponding ketone (0.168 mmol, 1.2 eq.). The requisite aldehyde (0.14 mmol, 1 eq.) was added to the reaction mixture. The resulting mixture was stirred at room temperature for 24 h and then treated with saturated aqueous ammonium chloride. The mixture was extracted with ethyl acetate (3 × 2 mL). The combined organic extract was washed with brine, dried over Na2SO4, and concentrated in vacuo. NMR analysis determined the diastereomeric ratio, and the residue was purified by flash column chromatography with hexane/ethyl acetate (3:1) to produce the aldol products that were subjected to chiral HPLC analysis to determine enantiomeric excesses.
5. Conclusions
Several new peptide catalysts which mimic based on promiscuous enzyme AKRs were designed and synthesized, and proved to be excellent multifunctional organocatalysts for direct asymmetric aldol reactions. High enantioselectivities were observed for most of the aldol reactions. Direct aldol reactions between selected ketones and aromatic aldehydes indicated a very impressive selectivity over a range of direct aldol reactions. The present study offers a proof-of-principle and indicates that proline/glutamic acid and hystidine lysine as a hydrophilic residue and leucine, phenylalanine, valine as hydrophobic residues in 8aa have significant potentials in catalysis. The carboxyl group of glutamic acid, lysine and histidine provided an obvious site for facile attachment of structural units, which can provide alternate hydrogen bonding arrays. The secondary structures of the peptides were studied using CD spectra and FT-IR. 8aa has the advantage of functioning as a powerful asymmetric organocatalyst in aqueous medium and of being reusable.References
- V. Bisai, A. Bisai and V. K. Singh, Enantioselective organocatalytic aldol reaction using small organic molecules, Tetrahedron, 2012, 68(24), 4541–4580 CrossRef CAS PubMed.
- M. Nielsen, D. Worgull, T. Zweifel, B. Gschwend, S. Bertelsen and K. A. Jørgensen, Mechanisms in aminocatalysis, Chem. Commun., 2011, 47(2), 632–649 RSC.
- M. M. Heravi and S. Asadi, Recent applications of organocatalysts in asymmetric aldol reactions, Tetrahedron: Asymmetry, 2012, 23(20), 1431–1465 CrossRef CAS PubMed.
- R. Mahrwald and D. Evans, Modern aldol reactions, Wiley-VCH, Weinheim, vol. 2, 2004 Search PubMed.
- P. I. Dalko and L. Moisan, In the golden age of organocatalysis, Angew. Chem., 2004, 43(39), 5138–5175 CrossRef CAS PubMed.
- B. List, R. A. Lerner and C. F. Barbas, Proline-catalyzed direct asymmetric aldol reactions, J. Am. Chem. Soc., 2000, 122(10), 2395–2396 CrossRef CAS.
- Z. G. Hajos and D. R. Parrish, Asymmetric synthesis of bicyclic intermediates of natural product chemistry, J. Org. Chem., 1974, 39(12), 1615–1621 CrossRef CAS.
- H. Wennemers, Asymmetric catalysis with peptides, Chem. Commun., 2011, 47(44), 12036–12041 RSC.
- H. Wennemers, Peptides as asymmetric catalysts and templates for the controlled formation of Ag nanoparticles, J. Pept. Sci., 2012, 18(7), 437–441 CrossRef CAS PubMed.
- S. B. Ötvös, I. M. Mándity and F. Fülöp, Asymmetric aldol reaction in a continuous-flow reactor catalyzed by a highly reusable heterogeneous peptide, J. Catal., 2012, 295, 179–185 CrossRef PubMed.
- K. Akagawa and K. Kudo, Construction of an All-Carbon Quaternary Stereocenter by the Peptide-Catalyzed Asymmetric Michael Addition of Nitromethane to β-Disubstituted α, β-Unsaturated Aldehydes, Angew. Chem., Int. Ed., 2012, 51(51), 12786–12789 CrossRef CAS PubMed.
- K. Kudo, K. Akagawa and R. Suzuki, Effect of the Helical Tether of a Resin-Supported Peptide Catalyst for Friedel–Crafts-Type Alkylation in Water, Adv. Synth. Catal., 2012, 354(7), 1280–1286 CrossRef PubMed.
- G. Szőllősi, A. Csámpai, C. Somlai, M. Fekete and M. Bartók, Unusual enantioselectivities in heterogeneous organocatalyzed reactions: Reversal of direction using proline di- versus tri-peptides in the aldol addition, J. Mol. Catal. A: Chem., 2014, 382(0), 86–92 CrossRef PubMed.
- S. Bahmanyar, K. Houk, H. J. Martin and B. List, Quantum mechanical predictions of the stereoselectivities of proline-catalyzed asymmetric intermolecular aldol reactions, J. Am. Chem. Soc., 2003, 125(9), 2475–2479 CrossRef CAS PubMed.
- B. List, Proline-catalyzed asymmetric reactions, Tetrahedron, 2002, 58(28), 5573–5590 CrossRef CAS.
- J. Kofoed, J. Nielsen and J.-L. Reymond, Discovery of new peptide-based catalysts for the direct asymmetric aldol reaction, Bioorg. Med. Chem. Lett., 2003, 13(15), 2445–2447 CrossRef CAS.
- W. Zou, I. Ibrahem, P. Dziedzic, H. Sundén and A. Córdova, Small peptides as modular catalysts for the direct asymmetric aldol reaction: ancient peptides with aldolase enzyme activity, Chem. Commun., 2005,(39), 4946–4948 RSC.
- W. Huang, H. Tian, H. Xu, L. Zheng, Q. Liu and S. Zhang, L-Valine dipeptide organocatalysts with two amide units for the direct asymmetric aldol reaction in brine, Catal. Lett., 2011, 141(6), 872–876 CrossRef CAS.
- P. Dziedzic, W. Zou, J. Háfren and A. Córdova, The small peptide-catalyzed direct asymmetric aldol reaction in water, Org. Biomol. Chem., 2006, 4(1), 38–40 CAS.
- A. Córdova, W. Zou, P. Dziedzic, I. Ibrahem, E. Reyes and Y. Xu, Direct asymmetric intermolecular aldol reactions catalyzed by amino acids and small peptides, Chem.–Eur. J., 2006, 12(20), 5383–5397 CrossRef PubMed.
- E. Bellis and G. Kokotos, Proline-modified poly(propyleneimine) dendrimers as catalysts for asymmetric aldol reactions, J. Mol. Catal. A: Chem., 2005, 241(1–2), 166–174 CrossRef CAS PubMed.
- J. Gao, J. Liu, D. Jiang, B. Xiao and Q. Yang, l-Prolinamide functionalized mesoporous silicas: Synthesis and catalytic performance in direct aldol reaction, J. Mol. Catal. A: Chem., 2009, 313(1–2), 79–87 CrossRef CAS PubMed.
- P. Krattiger, R. Kovasy, J. D. Revell, S. Ivan and H. Wennemers, Increased structural complexity leads to higher activity: peptides as efficient and versatile catalysts for asymmetric aldol reactions, Org. Lett., 2005, 7(6), 1101–1103 CrossRef CAS PubMed.
- J. D. Revell, D. Gantenbein, P. Krattiger and H. Wennemers, Solid-supported and pegylated H–Pro–Pro–Asp–NHR as catalysts for asymmetric aldol reactions, Pept. Sci., 2006, 84(1), 105–113 CrossRef CAS PubMed.
- J. D. Revell and H. Wennemers, Functional group requirements within the peptide H-Pro-Pro-Asp-NH2 as a catalyst for aldol reactions, Tetrahedron, 2007, 63(35), 8420–8424 CrossRef CAS PubMed.
- J. D. Revell and H. Wennemers, Peptidic catalysts developed by combinatorial screening methods, Curr. Opin. Chem. Biol., 2007, 11(3), 269–278 CrossRef CAS PubMed.
- E. A. Davie, S. M. Mennen, Y. Xu and S. J. Miller, Asymmetric catalysis mediated by synthetic peptides, Chem. Rev., 2007, 107(12), 5759–5812 CrossRef PubMed.
- B. R. Linton, M. H. Reutershan, C. M. Aderman, E. A. Richardson, K. R. Brownell, C. W. Ashley, C. A. Evans and S. J. Miller, Asymmetric Michael addition of α-nitro-ketones using catalytic peptides, Tetrahedron Lett., 2007, 48(11), 1993–1997 CrossRef CAS PubMed.
- S. J. Miller, In search of peptide-based catalysts for asymmetric organic synthesis, Acc. Chem. Res., 2004, 37(8), 601–610 CrossRef CAS PubMed.
- F. Chen, S. Huang, H. Zhang, F. Liu and Y. Peng, Proline-based dipeptides with two amide units as organocatalyst for the asymmetric aldol reaction of cyclohexanone with aldehydes, Tetrahedron, 2008, 64(40), 9585–9591 CrossRef CAS PubMed.
- C. Andreu and G. Asensio, Effect of addition of Lewis/Brönsted acids in the asymmetric aldol condensation catalyzed by trifluoroacetate salts of proline-based dipeptides, Tetrahedron, 2012, 68, 7966–7972 CrossRef CAS PubMed.
- D. Zhao and K. Ding, Recent Advances in Asymmetric Catalysis in Flow, ACS Catal., 2013, 3(5), 928–944 CrossRef CAS.
- M. Wiesner, J. D. Revell and H. Wennemers, Tripeptides as Efficient Asymmetric Catalysts for 1,4-Addition Reactions of Aldehydes to Nitroolefins–A Rational Approach, Angew. Chem., Int. Ed., 2008, 47(10), 1871–1874 CrossRef CAS PubMed.
- X. Y. Gong, D. Dobrunz, M. Kümin, M. Wiesner, J. D. Revell, H. Wennemers and P. C. Hauser, Separating stereoisomers of di-, tri-, and tetrapeptides using capillary electrophoresis with contactless conductivity detection, J. Sep. Sci., 2008, 31(3), 565–573 CrossRef CAS PubMed.
- S. Bayat, B. A. Tejo, A. B. Salleh, E. Abdmalek, Y. M. Normi and M. B. A. Rahman, Various Polar Tripeptides as Asymmetric Organocatalyst in Direct Aldol Reactions in Aqueous Media, Chirality, 2013, 25(11), 726–734 CrossRef CAS PubMed.
- S. S. Hoog, J. E. Pawlowski, P. M. Alzari, T. M. Penning and M. Lewis, Three-dimensional structure of rat liver 3 alpha-hydroxysteroid/dihydrodiol dehydrogenase: a member of the aldo-keto reductase superfamily, Proc. Natl. Acad. Sci. U. S. A., 1994, 91(7), 2517–2521 CrossRef CAS.
- J. Jez, M. Bennett, B. Schlegel, M. Lewis and T. Penning, Comparative anatomy of the aldo–keto reductase superfamily, Biochem. J., 1997, 326, 625–636 CAS.
- S. D. Copley, Enzymes with extra talents: moonlighting functions and catalytic promiscuity, Curr. Opin. Chem. Biol., 2003, 7(2), 265–272 CrossRef CAS.
- C.-J. Li and L. Chen, Organic chemistry in water, Chem. Soc. Rev., 2006, 35(1), 68–82 RSC.
- C. Allemann, J. M. Um and K. N. Houk, Computational investigations of the stereoselectivities of proline-related catalysts for aldol reactions, J. Mol. Catal. A: Chem., 2010, 324(1–2), 31–38 CrossRef CAS PubMed.
- X. Q. Yu, Z. B. Xie, N. Wang and G. F. Jiang, Biocatalytic asymmetric aldol reaction in buffer solution, Tetrahedron Lett., 2013, 54(8), 945–948 CrossRef PubMed.
- N. K. Rana, R. Unhale and V. K. Singh, Enantioselective sulfa-Michael addition of thioacids to α,β-unsaturated ketones with bifunctional organocatalyst, Tetrahedron Lett., 2012, 53(16), 2121–2124 CrossRef CAS PubMed.
- J. G. Hernández and E. Juaristi, Efficient ball-mill procedure in the ‘green’asymmetric aldol reaction organocatalyzed by-proline-containing dipeptides in the presence of water, Tetrahedron, 2011, 67(36), 6953–6959 CrossRef PubMed.
- K. N. Houk and B. List, Asymmetric organocatalysis, Acc. Chem. Res., 2004, 37(8), 487 CrossRef CAS.
- E. Saguer, P. Alvarez and A. A. Ismail, Heat-induced denaturation/aggregation of porcine plasma and its fractions studied by FTIR spectroscopy, Food Hydrocolloids, 2012, 27(1), 208–219 CrossRef CAS PubMed.
- E. Goormaghtigh, J.-M. Ruysschaert and V. Raussens, Evaluation of the information content in infrared spectra for protein secondary structure determination, Biophys. J., 2006, 90(8), 2946–2957 CrossRef CAS PubMed.
- J. Alsina, C. Chiva, M. Ortiz, F. Rabanal, E. Giralt and F. Albericio, Active carbonate resins for solid-phase synthesis through the anchoring of a hydroxyl function. Synthesis of cyclic and alcohol peptides, Tetrahedron Lett., 1997, 38(5), 883–886 CrossRef CAS.
- F. Albericio, Developments in peptide and amide synthesis, Curr. Opin. Chem. Biol., 2004, 8(3), 211–221 CrossRef CAS PubMed.
- P. I. Haris and D. Chapman, The conformational analysis of peptides using Fourier transform IR spectroscopy, Biopolymers, 1995, 37(4), 251–263 CrossRef CAS PubMed.
- W. K. Surewicz, H. H. Mantsch and D. Chapman, Determination of protein secondary structure by Fourier transform infrared spectroscopy: a critical assessment, Biochemistry, 1993, 32(2), 389–394 CrossRef CAS.
- J. Kong and S. Yu, Fourier transform infrared spectroscopic analysis of protein secondary structures, Acta Biochim. Biophys. Sin., 2007, 39(8), 549–559 CrossRef CAS PubMed.
- R. B. Merrifield, Solid phase peptide synthesis. I. The synthesis of a tetrapeptide, J. Am. Chem. Soc., 1963, 85(14), 2149–2154 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra04866k |
| This journal is © The Royal Society of Chemistry 2014 |