Trienediynes on a 1,3,5-trisubstituted benzene template: a new approach for enhancement of reactivity†
Ishita Hatiala,
Saibal Janaa,
Shrabani Bisaia,
Manasmita Dasb,
Ananta Kumar Ghoshc,
Anakuthil Anoop*a and
Amit Basak*a
aIndian Institute of Technology Kharagpur, Chemistry, Kharagpur, India. E-mail: absk@chem.iitkgp.ernet.in
bUniversity of North Carolina, Chapel Hill, Neurology, US
cIndian Institute of Technology Kharagpur, Biotechnology, Kharagpur, India
First published on 13th June 2014
Abstract
The synthesis of trienediynes based on a 1,3,5-trisubstituted benzene template is described. The presence of three adjacent enediyne moieties in the dendritic core exerted a cooperative effect to bring down the onset temperature for Bergman cyclization leading to increased DNA cleavage in a shorter time.
An important aspect of the design and development of antineoplastic drugs is to enhance the efficiency of cleavage of the genetic material. Naturally occurring enediynes and their synthetic analogues1 are very potent cytotoxins; this extreme potency makes them attractive for their use as effective prodrugs.2,3 The cytotoxic effects of these compounds are triggered by molecular rearrangements which bring the conjugated triple bonds of the enediyne core sufficiently close to initiate an electrocyclic reaction known as Bergman cyclization (BC).4 Formation of a transient benzene 1,4-diradical capable of simultaneous abstraction of a proton (at C-4′ or C5′) from a ribose moiety on each DNA chain results in a cascade of reactions including a final oxidative cleavage.5 The cleavage process being radical mediated, the efficiency of cleavage is dependent upon the kinetics and onset temperature for BC. In order to develop a DNA cleavage-based antitumor agent, one needs to consider the cleavage dynamics which in turn depends upon several factors like internal strain present in a cyclic system,6 chelation,7 pH changes,8 intramolecular H-bond,9 ortho effect10 etc. Many of these have been exploited in the design of several artificial enediynes; however, to our knowledge, so far there is no report on the reactivity of enediynes when two or more of them are held close-by on a template. In these cases, it is possible that their mutual electronic and or steric interactions may influence their reactivity towards BC. The situation is schematically presented in Fig. 1.
 | ||
| Fig. 1 Rationale behind the template-based trienediyne design. | ||
To have an idea on the effect of keeping three enediyne units on a template upon its reactivity towards BC, we carried out geometry optimization at BP86-D3/def2-SVP11 level of theory using Orca-2.9.1 (ref. 12) on some of these enediyne systems. We were particularly interested to know the distance between the acetylenic ends (c and d-distance) and the dihedral angle between the benzene ring and the attached amide carbonyl. The calculated results are shown in Table 1. Interestingly, the c and d-distance in one of the enediyne unit in the trimeric system came out to be lower as compared to that in the monomeric counterpart. This will activate at least one of the enediyne unit towards diradical formation via BC. The other interesting feature is the greated dihedral angle between the planes, one containing benzene ring and the other the carbonyl. Higher dihedral angle means lower conjugation of the carbonyl with the benzene π-cloud and greater conjugation with the enediyne amide N. These two factors lower c and d-distance and greater conjugation of amide N are expected to make the trienediyne more reactive. The energy minimized structures are shown in Fig. 2.
| Dendrimer | Dihedral angle (°) | Distance (Å) |
|---|---|---|
| Monomer | 1–2–3–4 = 40.747 | 5–6 = 3.37671 |
| Trimer | 1–2–3–4 = 50.879 | 5–6 = 3.22898 |
| 7–8–9–10 = 35.702 | 11–12 = 3.37951 | |
| 13–14–15–4 = 47.611 | 16–17 = 3.37748 | |
| Monomer | 1–2–3–4 = 42.193 | 5–6 = 3.35003 |
| Trimer | 1–2–3–4 = 47.704 | 5–6 = 3.20855 |
| 7–8–9–10 = 41.350 | 11–12 = 3.35616 | |
| 13–14–15–4 = 54.436 | 16–17 = 3.35614 |
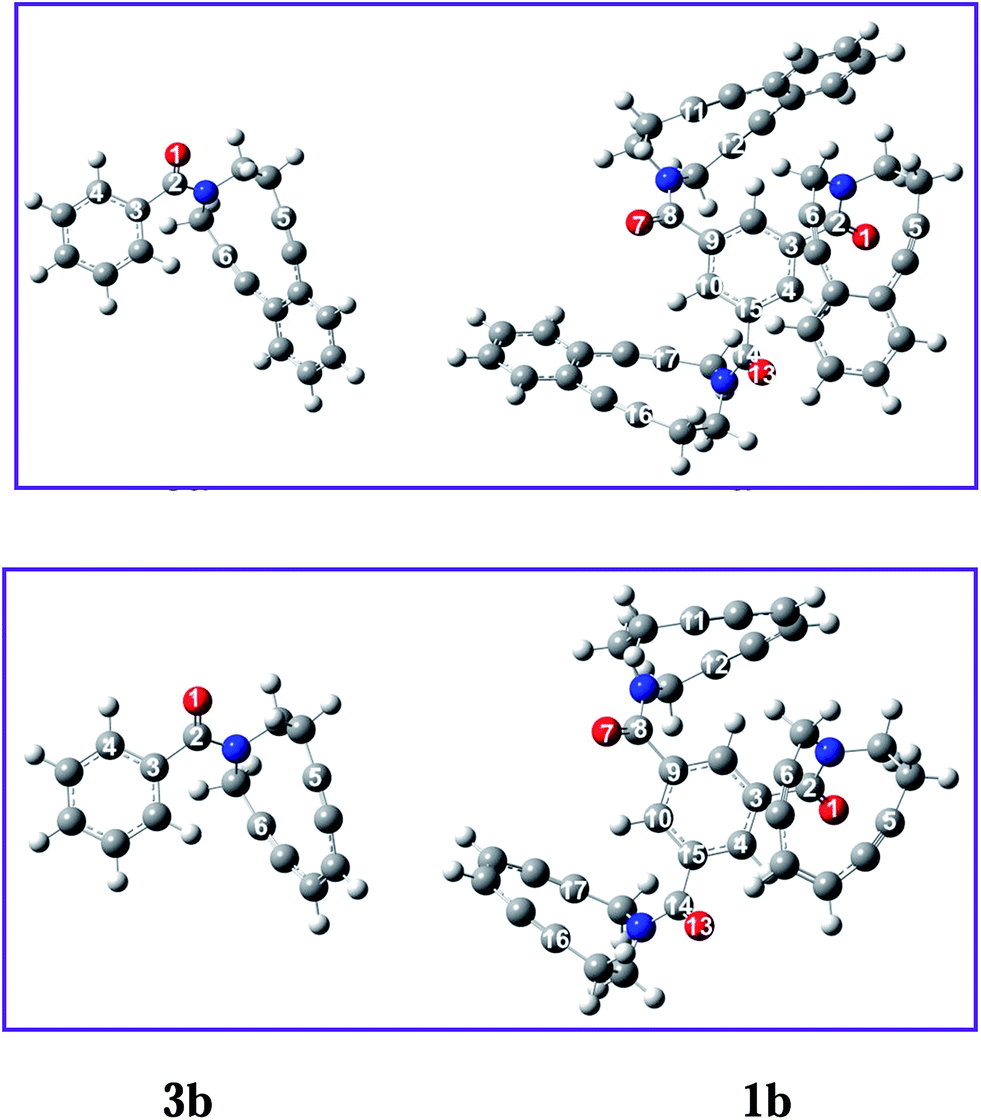 | ||
| Fig. 2 The energy minimized structures of mono and trienediynes. | ||
With the backing of computational results, we went ahead and synthesized three types of trienediyne 1a–c, based on a dendritic core 1,3,5-benzenetricarbonyl chloride (trimesoyl chloride) (Fig. 3). These trienediynes were found to be considerably more reactive than their monomeric counterpart 3a–c. The synthesis and reactivity profile of the trienediynes are described here. The comparative study on the DNA-cleavage activity of one trienediyne vis-à-vis the monocyclic counterpart is also described.
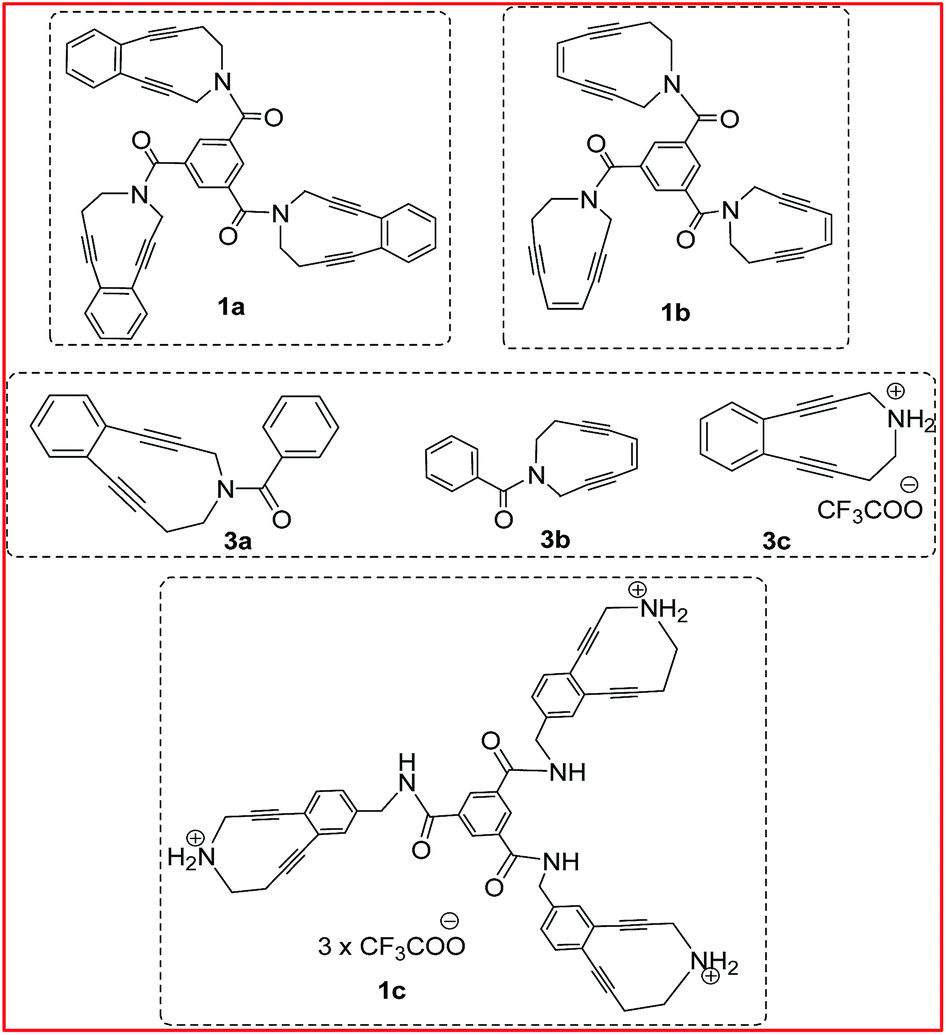 | ||
| Fig. 3 The designed trienediynes and the monomeric counterparts. | ||
The synthesis of the trienediyne is quite straight-forward. The enediynes 1a and 1b were prepared from the free amines13 2a and 2b via acylation with trimesoyl chloride. These were purified by chromatography over silica gel. Extreme care had to be taken for the synthesis of aliphatic trienediyne 1b because of its low half-life under ambient conditions. For the synthesis of 1c, the N-nosyl enediynyl benzyl amine was first prepared from the corresponding Boc-protected derivative. The latter was the coupled with trimesoyl chloride which was followed by deprotection with thiophenol/K2CO3 to provide the free amine, isolated as the trifluoro acetate salt 1c (Scheme 1).
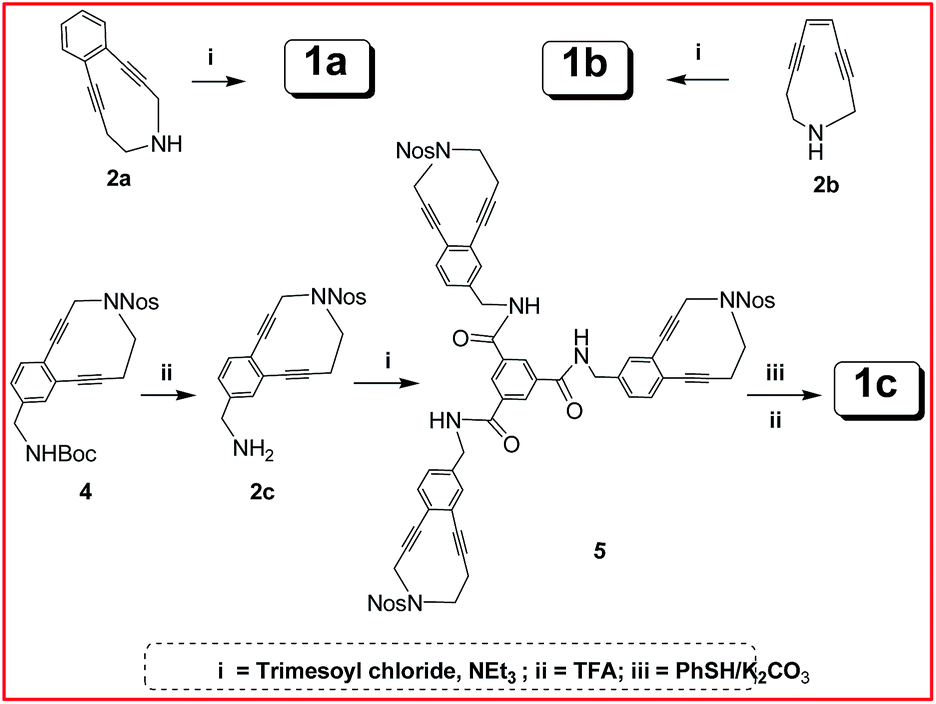 | ||
| Scheme 1 Synthesis of trienediynes. | ||
With all substrates in hand, we proceeded to determine the thermal reactivity of the trienediynes 1a–c under neat conditions by DSC (Fig. 4). These trienediynes showed higher reactivity compared to monomeric counterparts as reflected in the onset temperature for BC (Table 2).14 The difference in reactivity is most striking in case of 1b and 1c.
 | ||
| Fig. 4 DSC thermograms of various enediynes (the arrows indicate onset temperatures). | ||
| Substrate | Onset temp. for BC | Substrate | Onset temp. for BC | ΔTonset |
|---|---|---|---|---|
| 1a | 85 °C | 3a | 100 °C | 15 °C |
| 1b | 35 °C | 3b | 60 °C | 25 °C |
| 1c | 75 °C | 3c | 155 °C | 80 °C |
| 5 | 130 °C | 4 | 150 °C | 20 °C |
In order to resolve the issue solution phase reactivity, a DMSO solution of 1c (0.005 μM) was kept in a thermostatic bath, fixed at a preset temperature and by taking the NMR at different time points, an effort was made to study the precise kinetics of Bergman cyclization. In solution phase, the molecule started to cyclize only when heated to 75 °C and above. In the 1H NMR spectra, the signals for the methylene protons diminished over time, while new peaks for the cycloaromatised product in the aromatic region and between δ 4–5 started to appear.
However, the cycloaromatization of three enediyne units centering the common adaptor in a single molecule complicated the overall situation due to overlapping of signals and that made evaluation of kinetic parameters on the basis of integration values rather difficult. The matter was finally resolved by high performance liquid chromatographic (HPLC) analysis. The latter involved heating a solution of 1a in CHCl3 in a sealed tube at 75 °C containing an excess of 1,4-cyclohexadiene and naphthalene (used as internal standard). A definite amount of aliquot was taken from the reaction mixture at regular time intervals and injected for HPLC analysis. The disappearance of the starting material with time, followed by concomitant appearance of new peaks corresponding to the cycloaromatized products could be clearly envisioned from the HPLC chromatogram. Both HPLC as well as NMR spectroscopic data however suggests that kinetics of Bergman cyclization in a dendritic system is highly complicated and falls in the domain of consecutive reactions. Cycloaromatization of the three enediyne units is most likely to occur in successive steps, every step having its own reaction rate. However, the rate of disappearance of starting enediyne against the reference (in this case naphthalene) may be taken as a reflection of the rate of first BC of one amongst the three enediyne units. It was found that the rate of disappearance of 1a followed a first order kinetics (k = 1.2 × 10−4 s−1 at 75 °C) with a significantly short half life of 1.6 hours. The experiment was repeated with other endediynes 3a, 1c and 3c (Table 3). The BC of monoenediyne 3a is about 11.4 times slower (k = 1.05 × 10−5 s−1 at 75 °C). For the other pair 1c and 3c, the trienediyne 1c is 7.6 times faster as compared to that for the corresponding mono enediyne 3c thus confirming dendritic amplification on the rate of Bergman cyclization.
| Substrate | Rate constant (k) | Half life (t1/2) at 75 °C |
|---|---|---|
| 1a | 1.2 × 10−4 s−1 | 1.6 h |
| 3a | 1.05 × 10−5 s−1 | 18.2 h |
| 1c | 1.8 × 10−4 s−1 | 1.0 h |
| 3c | 2.5 × 10−5 s−1 | 7.7 h |
The enhancement of kinetics of BC for the trimeric enediyne 1b as compared to the monomeric counterpart 3b prompted us to compare their DNA cleavage activity. We expected that 3b should cleave DNA more rapidly. Thus a solution of 3b and 1b in DMSO was incubated with supercoiled plasmid DNA pBR322. Because of the presence of three enediyne units, the concentration of the trienediyne 1b has been maintained at 1/3rd of that of the monocyclic counterpart 3b. The appearance of Form II (∼15%) could be seen after 6 h for the trienediyne 1b (lane III in Fig. 5A). The cleavage pattern is almost similar after 24 h of incubation (Fig. 5B). At similar concentrations, the trienediyne expectedly showed faster DNA cleavage (Fig. 5C and D). The effect is more dramatic in case of the pair 1c/3c. In this case the cleavage is slow but it is clear from the gel picture that after 30 h, the trienediyne 1c has induced greater formation of nicked form (FII) as compared to the monoenediyne 3c (Fig. 6).
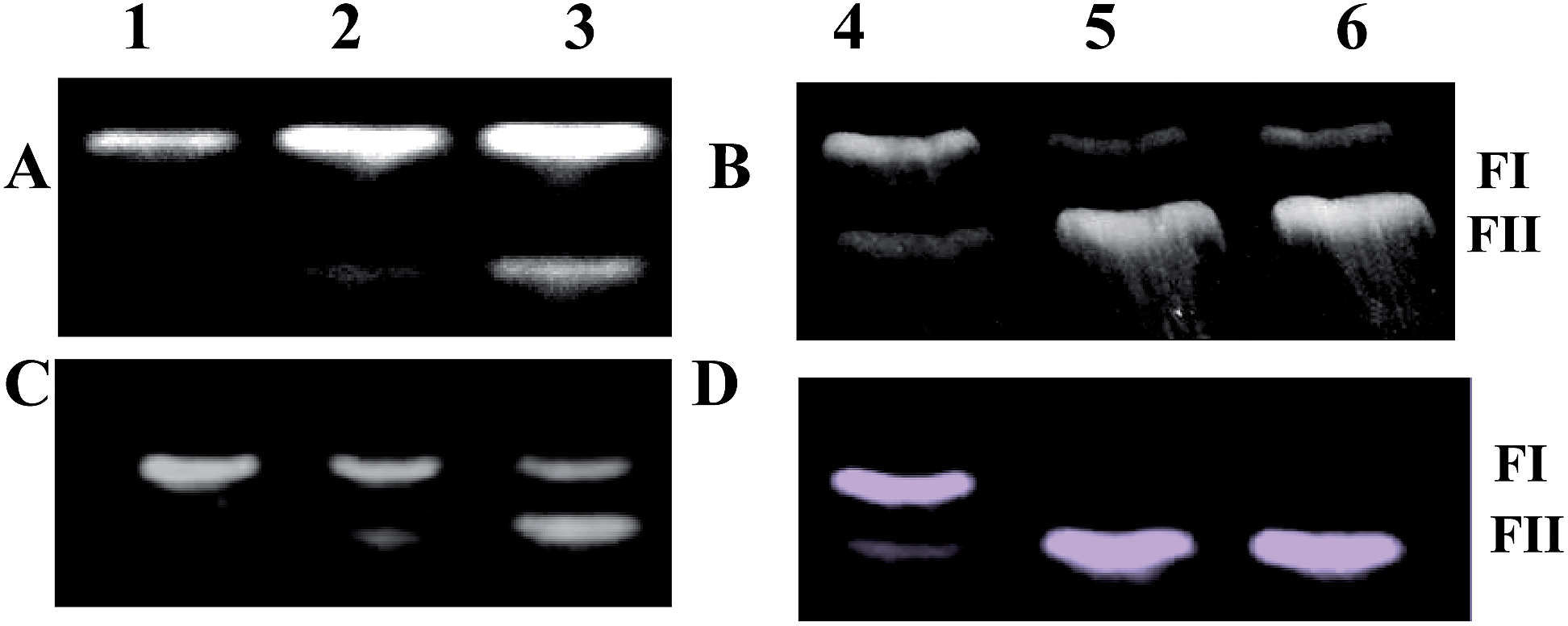 | ||
| Fig. 5 For A and B: plasmid relaxation assays with compounds 1b and 3b (5 μL each from a stock of 20 μM (for 3b) and 7 μM (for 1b) in DMSO) and pBR 322 plasmid DNA (7 μL from a stock of 0.03 μg μL−1 at pH 8.0). For C and D: (5 μL each from a stock of 20 μM in DMSO) and pBR 322 DNA (7 μL from a stock of 0.03 μg μL−1 at pH 8.0). These were separately mixed with 20 mM phosphate buffer of pH 7.5, incubated at 37 °C for 6 h in A and C and for 24 h in B and D; lanes 1 & 4: DNA alone, 2 & 5: DNA with 3b, 3 & 6: DNA with 1b. | ||
 | ||
| Fig. 6 Plasmid relaxation assays with compounds 1c and 3c (5 μL each from a stock of 30 μM (for 3c) and 10 μM (for 1c) in DMSO) and pBR 322 plasmid DNA (7 μL from a stock of 0.03 μg μL−1 at pH 6.5). These were separately mixed with 20 mM phosphate buffer of pH 6.5, incubated at 37 °C for 30 h in A; lanes 1: DNA alone, 2: DNA with 3c, 3: DNA with 1c. | ||
Thus we have demonstrated that trimeric enediynes built on a dendriting 1,3,5-benzene core showed higher reactivity towards BC and hence faster DNA cleavage. These results have provided a new strategy for enediyne activation. Current studies are aimed towards synthesizing higher generation enediyne dendrimers and study their thermal and biological reactivities.
Acknowledgements
DST is acknowledged for the J C Bose Fellowship and a research grant grant to AB, which supported the research. IH and SJ are grateful to CSIR and UGC, Government of India, respectively, for research fellowships. The NMR facility has been provided at IIT Kharagpur by DST under the IRPHA programme.References
- For review, see: (a) R. K. Mohamed, P. W. Peterson and I. V. Alabugin, Chem. Rev., 2013, 113, 7089 CrossRef CAS PubMed; (b) D. Rawat and J. M. Zaleski, Synlett, 2004, 0393 CAS; (c) I. A. Maretina and B. A. Trofimov, Russ. Chem. Rev., 2006, 75, 825 CrossRef CAS PubMed; (d) W.-Y. Yang, B. Breiner, S. V. Kovalenko, C. Ben, M. Singh, S. N. LeGrand, Q.-X. A. Sang, G. F. Strouse, J. A. Copland and I. V. Alabugin, J. Am. Chem. Soc., 2009, 131, 11458 CrossRef CAS PubMed; (e) M. C. Joshi and D. S. Rawat, Chem. Biodiversity, 2012, 9, 459 CrossRef CAS PubMed; (f) S. V. Kovalenko and I. V. Alabugin, Chem. Commun., 2005, 1444 RSC.
- (a) S. C. Sinha, L.-S. Li, G. P. Miller, S. Dutta, C. S. Rader and R. A. Lerner, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 3095 CrossRef CAS PubMed; (b) K. C. Nicolaou, W. M. Dai, S. C. Tsay, V. A. Estevez and W. Wrasidlo, Science, 1992, 256, 117 CrossRef; (c) K. C. Nicolaou, G. Zuccarello, Y. Ogawa, E. J. Schweiger and T. Kumazawa, J. Am. Chem. Soc., 1988, 110, 4866 CrossRef CAS; (d) B. Breiner, K. Kaya, S. Roy, W.-Y. Yang and I. V. Alabugin, Org. Biomol. Chem., 2012, 10, 3974 RSC; (e) B. Breiner, J. C. Schlatterer, I. V. Alabugin, S. V. Kovalenko and N. L. Greenbaum, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 13016 CrossRef CAS PubMed.
- (a) P. R. Schreiner, J. Am. Chem. Soc., 1998, 120, 4184 CrossRef CAS; (b) P. R. Schreiner, J. Chem. Soc. Chem. Commun., 1998, 483 RSC.
- (a) R. P. Jones and R. G. Bergman, J. Am. Chem. Soc., 1972, 94, 660 CrossRef CAS; (b) R. G. Bergman, Acc. Chem. Res., 1973, 6, 25 CrossRef CAS; (c) T. P. Lockhart and R. G. Bergman, J. Am. Chem. Soc., 1981, 103, 4091 CrossRef CAS.
- (a) W. M. Dai and K. C. Nicolaou, Angew. Chem., Int. Ed. Engl., 1991, 30, 1387 CrossRef; (b) W. M. Dai and K. C. Nicolaou, Angew. Chem., 1991, 103, 1453 CrossRef.
- A. Basak, U. K. Khamrai, U. K. Mullick, L. Banfi and G. Guanti, Chem. Commun., 1996, 749 (Angew. Chem., Int. Ed. Engl., 1995, 34, 2393) RSC.
- A. Basak, S. Mandal and S. S. Bag, Chem. Rev., 2003, 103, 4077 CrossRef CAS PubMed.
- M. Kar and A. Basak, Chem. Rev., 2007, 107, 2861 CrossRef CAS PubMed.
- (a) S. Roy, S. S. Bag and A. Basak, Tetrahedron, 2012, 68, 8600 CrossRef CAS PubMed; (b) A. Basak, S. S. Bag and A. Basak, Bioorg. Med. Chem., 2005, 13, 4096 CrossRef CAS PubMed; (c) A. Basak, S. S. Bag and H. M. M. Bdour, Chem. Commun., 2003, 2614 RSC; (d) A. Basak, S. S. Bag and A. K. Das, Eur. J. Org. Chem., 2005, 1239 CrossRef CAS.
- T. A. Zeidan, S. V. Kovalenko, M. Manoharan and I. V. Alabugin, J. Org. Chem., 2006, 71, 962 CrossRef CAS PubMed.
- (a) A. D. Becke, Phys. Rev. A: At., Mol., Opt. Phys., 1988, 38, 3098 CrossRef CAS; (b) J. P. Perdew, Phys. Rev. B: Condens. Matter Mater. Phys., 1986, 33, 8822 CrossRef; (c) A. Schaefer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571 CrossRef CAS PubMed; (d) S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- Orca 2.9.1, developed by Frank Neese, Max Planck Institute for Bioinorganic Chemistry, Muelheim/Ruhr, Germany.
- (a) I. Hatial, P. S. Addy, A. K. Ghosh and A. Basak, Tetrahedron Lett., 2013, 54, 854 CrossRef CAS PubMed; (b) A. Basak and S. Mandal, Tetrahedron Lett., 2002, 43, 4241 CrossRef CAS; (c) A. Basak, S. Mandal, A. K. Das and V. Bertolasi, Bioorg. Med. Chem. Lett., 2002, 12, 873 CrossRef CAS.
- (a) B. Konig and H. Rutters, Tetrahedron Lett., 1994, 35, 3501 CrossRef; (b) A. Basak, S. S. Bag, P. A. Majumder, A. K. Das and V. Bertolasi, J. Org. Chem., 2004, 69, 6927 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra04794j |
| This journal is © The Royal Society of Chemistry 2014 |