 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The pros and cons of lignin valorisation in an integrated biorefinery
Zea Strassberger,
Stefania Tanase and
Gadi Rothenberg*
Van't Hoff Institute for Molecular Sciences, University of Amsterdam, Science Park 904, 1098 XH Amsterdam, The Netherlands. E-mail: g.rothenberg@uva.nl; Web: http://hims.uva.nl/hcsc Fax: +31 20 525 5604
First published on 12th June 2014
Abstract
This short critical review outlines possible scenarios for using lignin as a feedstock in a biorefinery environment. We first explain the position of biomass with respect to fossil carbon sources and the possibilities of substituting these in tomorrow's transportation fuels, energy, and chemicals sectors. Of these, the conversion of biomass to chemicals is, in our opinion, the most worthy. Focusing on lignin, we describe the four main processes for its industrial separation (the Sulfite, Soda, Kraft, and Organosolv processes). Then, we detail several short- and long-term perspectives for its valorisation to aromatics, polymers and materials, as well as new products and in-the-pipeline processes. Finally, we examine the limitations in current lignin valorisation and suggest possible ways forward. Combining the chemical aspects with up-to-date data from economic analyses gives a pragmatic and realistic overview of the commercial applications and possibilities for lignin in the coming decades, where biomass will join shale gas and crude oil as a valid and economical carbon source.
Introduction
Anyone dealing with energy, chemicals, and especially renewables and biomass will tell you that the past decade has been an interesting one. Much more so, perhaps, than the half-century that preceded it. The second half of the 20th century was dominated by crude oil as the raw material for energy, transportation, and chemicals. Technologies for processing crude oil have been developing since the 1860s, and today's refineries are highly integrated industrial plants.1,2 But even though we were reminded that fossil fuels will not last forever (cf. as early as 1956 Hubbert's pioneering work3) it was business as usual until the onset of the so-called “peak oil”.4–6 With the help of public opinion, and the increasing threat of global warming, governments began to look seriously for alternatives to fossil fuels.7 Renewables, and especially biodiesel8 and bioethanol,9 which are relatively easy to implement, featured high on governmental wish lists.And then, out of the blue, came the discovery of shale gas. In fact, it was not the discovery of shale gas per se as the development of its efficient and cheap extraction that made it such a game-changer.10 In less than five years, the availability of gas across the world has changed radically the energy market. Depending on which forecast you read, reserves are projected to last between 50–200 years,11 with the US becoming one of the main energy exporters in world. This astounding change will have large economical and political ramifications. Currently, energy in the US is six times cheaper when it comes from gas compared to diesel, and the political consequences of energy independence will be far-reaching.
But the gas bonanza should not stop renewables. Mankind has been given a temporary reprieve, but fossil fuels will run out, and they are not renewable on any human time scale. Moreover, the change in feedstock, from oil to gas, carries with it formidable chemical challenges and opportunities, as we will discuss here.
The key point is that we're changing the feedstock, but not the product. People still need the same energy, transportation fuels, and chemicals. But now these must come chiefly from C1 and C2 feeds, rather than from long alkanes. This will also redefine the role of biomass and its derivatives. Fig. 1 shows the relationship between the various feedstocks and the products. While it is true that a refinery can turn almost any carbon-containing feedstock into almost any product, some routes are much simpler (and therefore more economical) than others. As such, shale gas and natural gas are highly suited for energy production, crude oil is typically used for making transportation fuels and chemicals, and coal is used primarily for energy production (albeit far less efficiently than gas). The dashed curves in Fig. 1 denote the less efficient (but definitely doable) routes, such as producing diesel from coal via gasification and Fischer–Tropsch.
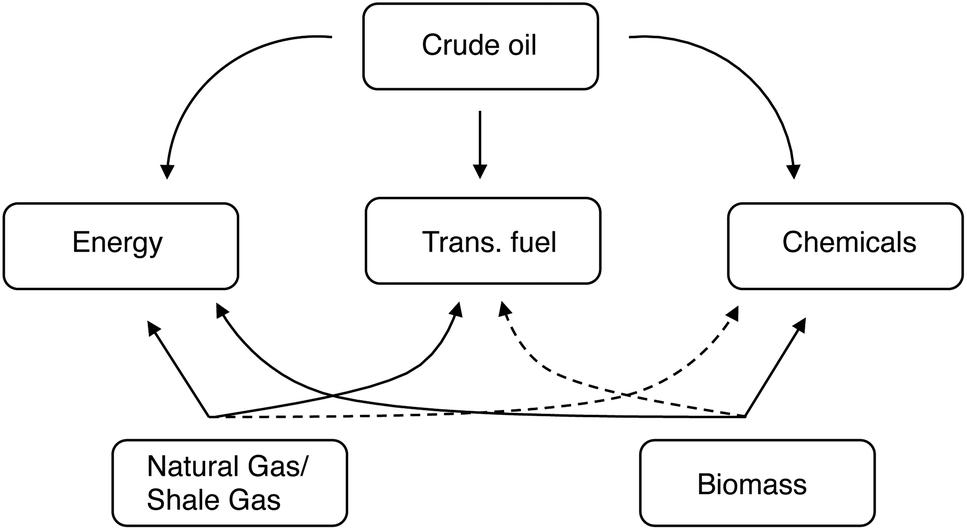 | ||
| Fig. 1 Feedstocks, routes, and products of the energy, transportation and chemicals sectors. The dashed curves represent less efficient connections. | ||
Now, let us consider the role that biomass can play in this scenario, taking into account the following five points:
• Gas is cheap and currently plentiful, but its transportation is costly.
• Biomass differs from crude oil and gas because it is over-functionalized and has a high oxygen content.
• Biomass is available practically worldwide, on varying scales.
• Unlike fossil hydrocarbons, biomass is renewable on a human timescale, but its large-scale cultivation raises ecological and economical concerns.
• Over 93 vol% of all the crude oil and gas processed today are converted into energy and fuels. All the bulk chemicals, polymers, fine-chemicals and pharmaceuticals account for less than 7%.
Based on these facts, we can see that it makes much more sense to convert biomass into chemicals than into energy. Since biomass is already highly functionalized, smart chemical conversion would allow us to use these functions. Moreover, biomass is a lower-grade fuel precisely because it is already functionalized, so converting it “back” to hydrocarbons in order to burn those is simply a double waste.
Vegetal biomass is comprised of cellulose, hemicellulose, and lignin. Current biorefinery concepts emphasize cellulose and hemicellulose, and these have been reviewed extensively.12,13 In this critical review, we will give only a short overview on cellulose and hemicellulose actual application on commercial scale. For clarity, this is not a comprehensive overview on how biomass can be converted into small molecules. Rather, it is a critical assessment of lignin conversion to useful chemicals, examining the possibilities of including lignin valorisation in future biorefineries.14
From biomass to upgraded value products
Biomass is a mixture of organic molecules, containing hydrogen, oxygen, nitrogen, phosphorous and sulfur atoms, plus small quantities of alkali-, alkaline-earth metals and heavy metals.15 We can consider lignocellulosic biomass as collection a fibrous cellulose backbones with a hemicellulose coating that are “glued” by lignin.Although biomass is annually renewable, it is still a limited resource.16 Today, the mean global land area per capita is only two soccer fields (2 ha; this includes non-arable areas such as Antarctica and the deserts17). Nevertheless, the European Environment Agency estimates that Europe's biomass production capacity could grow up to 300 Mtons by 2030.18 Similarly, the US Department of Energy estimates that sustainable biomass production in the US could reach 1.2 Btons per year by 2060, and this without compromising edible crops.19 This last point is essential. Biomass used to replace petro-based products should not compete with the food sectors. Notably, even though biomass is an attractive raw material, its use raises also important problems such as waste water treatment.
Much of the biomass that is currently considered waste will become a valuable feedstock. Hence, the main goal of tomorrow's biorefineries will be the processing of lignocellulosic biomass by recycling forest and agricultural waste.10 These refineries will produce biobased energy and high value added chemicals. However, they should be based on small and flexible units to overcome the problem of biomass transportation. Indeed, renewable feedstocks can be incorporated in higher-value-added commercial scale processes, as we show in the examples below. In principle, such processes can be integrated in an agro–biofuel–biomaterial–biopower cycle (Fig. 2), as shown by Ragauskas et al.20 The short examples below pertain all to cellulose/hemicellulose derivatives (carbohydrates). However, a complete solution would also have to include the processing of lignin, which is the main focus of this review.
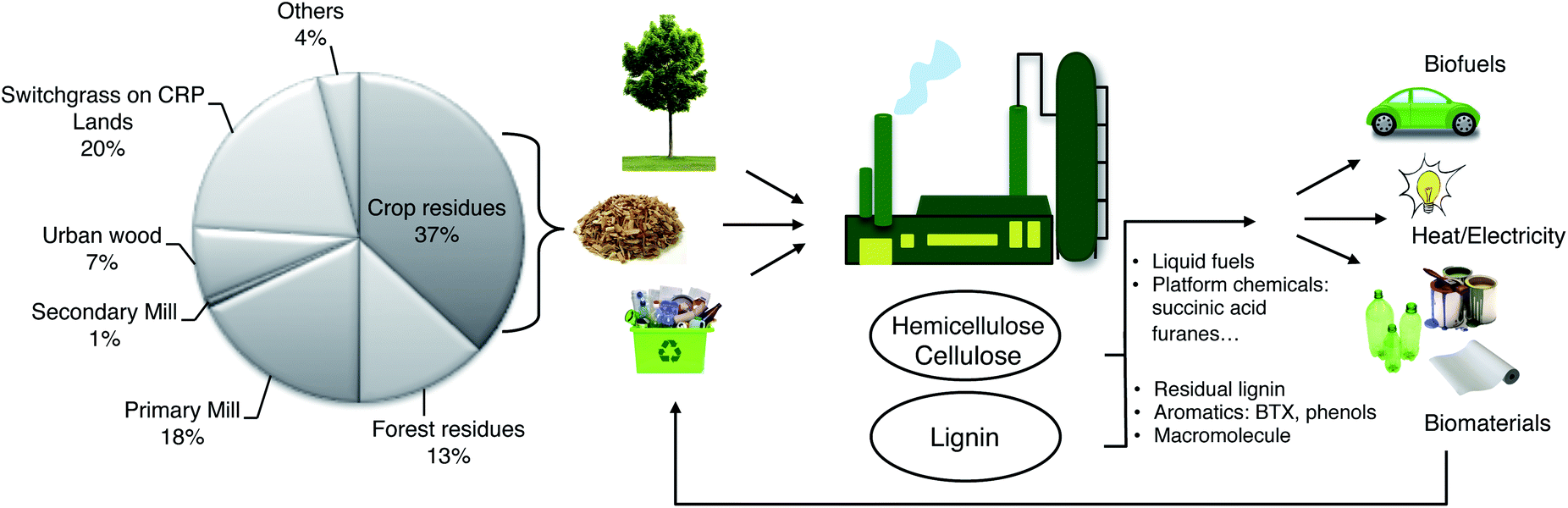 | ||
| Fig. 2 Cartoon showing the role of the future biorefinery as part of a fully integrated agro–biofuel–biomaterial–biopower cycle. The pie chart shows the distribution of vegetal biomass by volume. (CRP = Conservation Reserve Program). | ||
Current examples: bioplastics from cellulose and hemicellulose derivatives
Recent research has focused on the conversion of cellulose and hemicellulose, targeting the “top 12 platform chemicals” list put forward by the US Department of Energy (succinic acid, fumaric acid, malic acid, 2,5 furan dicarboxylic acid, aspartic acid, glutamic acid, levulinic acid, glycerol, sorbitol, arabinol and xylitol21–23). Note that these platform chemicals were also proposed by the US chemical industry. However, other important platform chemicals developed in EU were excluded. One promising market segment for these is the polymers industry. The global demand for conventional plastics in 2011 was 280 Mtons, of which 70% are made of polyolefins, PVC, PS, EPS or PET.24 As most of the petro-based plastics are not recycled, and not biodegradable, managing plastics waste is becoming one of mankind's biggest problems.25Biobased plastics (so-called “plantics”) are emerging slowly, but promisingly. Examples include the replacement of polyethene terephthalate (PET) with a biobased alternative that has similar chemical and physical properties, such as polylactic acid (PLA). This polymer can be blended with cellulose.26–28 Unlike PET, PLA is biodegradable. It can be recycled to its monomers by thermal depolymerization, or hydrolysis. Today, PLA is used in food packaging and the cosmetic industry. Natureworks, backed by Cargill, is the primary producer, with 140 ktpa.29 Similarly, polyhydroxyalkanoates (PHAs) are natural polyesters. Unlike PLA, they are UV stable as well as biodegradable. Industrially, PHAs are extracted and purified from bacteria.30 For example, Metabolix has commercialized under the trade name Mirel® a process leading to a broad range of PHAs biopolymers. In collaboration with Antibióticos in Spain, they announced a production of PHAs for 2013 of 10 ktpa.31
Another promising recent development is the YXY® process commercialised by Avantium Technologies in collaboration with Danone and the Cocoa-Cola company.21 YXY uses a chemocatalytic route to convert sugars into furan dicarboxylic acid (FDCA), a bio-replacement for terephthalic acid.32 The FDCA is then polymerized with ethanol to polyethylene furanoate (PEF). Coca Cola Company and Danone see this process as a route for developing biobased PEF bottles (whether FDCA would lead in practice to safer polymers will also depend on the toxicity profile of the resulting furanic derivatives).
Polyethylene itself can also be made from biomass, via biobased ethanol. Bioethanol is already widely produced. For example, POET, a commercial biorefinery in Lennox U.S., produces 54 ktpa.33 Once ethanol is obtained, the alcohol can be dehydrated to ethylene.34 Braskem is now the world leader in the field of biopolymers because of its Green PE (polyethylene), first produced on a commercial scale in September 2010 with a production capacity of 30 ktpa.35
But what applies to the nicely structured biomass fractions of cellulose and hemicellulose, does not necessarily apply also to lignin. A practical, if cynical, saying is “you can make anything from lignin, except money”.36 Here, we will try and understand the factors that can help unearth the now-hidden economical potential of lignin.37
Short- and long-term perspectives for lignin valorisation
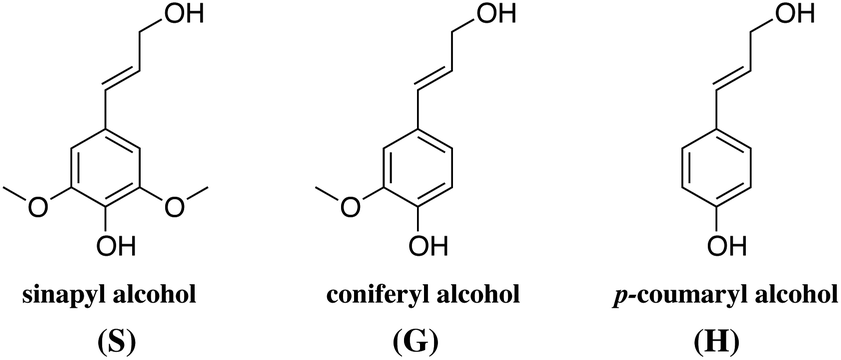 | ||
| Fig. 3 The three main monomer building blocks of lignin. | ||
The ratio between these units, the molecular weight and the amount of lignin differs from plant to plant. As a rule of thumb, the amount of lignin generally decreases from softwoods to hardwoods to grasses.40 The typical lignin content is 24–33% in softwoods, 19–28% in hardwoods depending on the sources, and 15–25% in cereal straws, bamboo or bagasse.40
Although the exact polymeric structure of lignin is undefined, the key substructures are known.39,41–43 The primary monomers of lignin are linked together through C–O bonds of α- and β-arylalkyl ethers.44,45 The β-O-4 linkage predominates, accounting for roughly 50% of the linkages.46 Other common linkages are the 5-5, β-5, and the dibenzodioxocin linkage (see Fig. 4). The ratio of linkages in softwood is given as an example in Table 1.
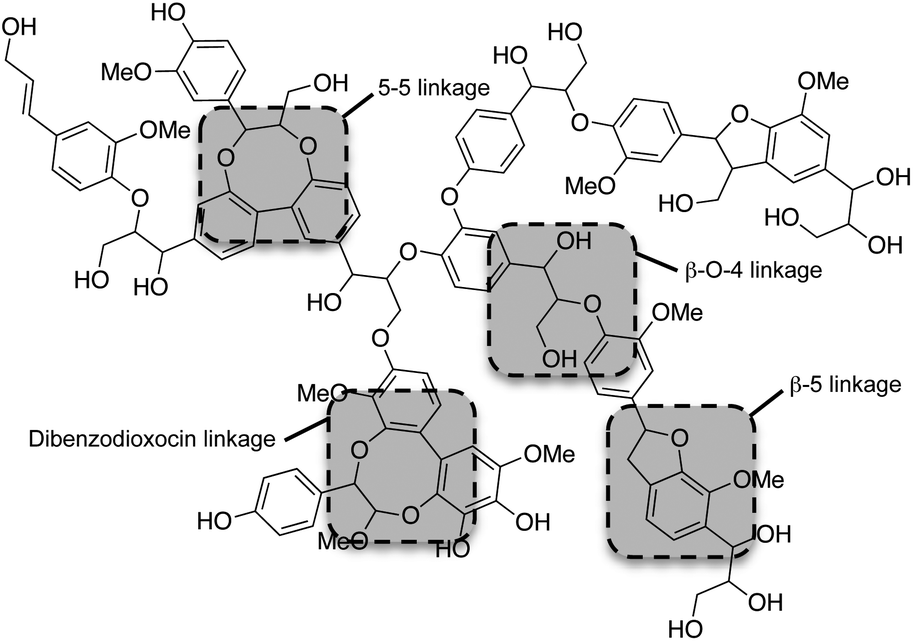 | ||
| Fig. 4 Schematic representation of lignin chemical bonds. | ||
| Linkages | Dimer structure | Distribution percentage [%] |
|---|---|---|
| β-O-4 | Phenylpropane β-aryl ether | 45−50 |
| 5-5 | Biphenyl and dibenzodioxocin | 18−25 |
| β-5 | Phenylcoumaran | 9−12 |
| α-O-4 | Phenylpropane α-aryl ether | 6−8 |
| β-1 | 1,2-Diaryl propane | 7−10 |
| 4-O-5 | Diaryl ether | 4−8 |
| β-β | β-β linked structures | 0–3 |
Using aqueous solution of sulfur dioxide with different pH, the Sulfite process is by far the largest producer of lignins (Table 2). The so-called lignosulfonates formed are water-soluble making it different from other technical lignins. As carbohydrates are not removed selectively, further purified lignins are obtained by removing the carbohydrate impurities by fermentation, chemical removal, ultrafiltration or selective precipitation.50
| Process | Scale (ktpa) | Sulfur | Suppliers |
|---|---|---|---|
| a Domtar's estimation for 2013.54b Pilot scale. | |||
| Sulfite (lignosulfonates) | ≈1000 | Yes | Borregaard LignoTech (NO, worldwide) |
| TEMBEC (FR, US) | |||
| Domjö Frabiker (SE) | |||
| La Rochette Venizel (FR) | |||
| NipponPaper chemicals (JNP) | |||
| Kraft | 60 | Yes | Meadwestvaco (US) |
| Kraft (LignoBoost) | 27a | Yes | Domtar (US) |
| Soda (sulfur free) | 5–10 | No | Greenvalue (CH, IND) |
| Organosolvb (sulfur free) | ≈3 | No | CIMV (FR) |
| Lignol innovations (CAN) | |||
| DECHEMA/Frauhoffer (DE) | |||
| Dedini (BR) | |||
The Soda lignin process uses sodium hydroxide to dissolve the lignin from lignocellulosic biomass.51 Lignin is recovered in several steps, including acid precipitation, maturation and filtration. This process gives sulfur-free lignin.
In the Kraft process, wood chips are boiled at 170 °C for about two hours in an aqueous solution of sodium hydroxide and sodium sulfide (so-called white liquor). During this treatment, the hydroxide and hydrosulfide anions depolymerize the lignin into smaller water/alkali-soluble fragments.45 The extraction of lignin in this process can be improved using the LignoBoost technology.52 LignoBoost works in conjunction with evaporation. By lowering the pH with CO2, lignin precipitates from the black liquor, dewatered using a filter press and then re-dissolved in water and acid. The resulting slurry is once again dewatered and washed, to produce a high purity lignin stream.53 In 2013, Domtar has successfully started up a 27 ktpa LignoBoost production plant.54
Finally, the Organosolv fractionation (also known as Alcell), with ethanol–water, yields high purity lignin with <1 wt% residual carbohydrate content.41 Organosolv pulping or fractionation of lignocellulosic biomass is one of the routes that can produce high-quality cellulose biofuel plus a high purity lignin. However, until now no organosolv process has reached commercial scale.55 It is possible to obtain technical lignin via other processes such as hydrolysis or steam explosion of biomass, but these are only at a pilot plant level (for availability of technical lignin see Table 2).
In 2010, the pulp and paper industry produced about 50 Mtons of low-purity lignin. Of this, only 2% was used commercially in the dispersants or binding sectors. The remaining 98% was recovered as a fuel.56 One further complication is that the structure of Kraft lignin, organosolv lignin, sulfite lignin and lignin differs between plant sources. Therefore, introducing lignin to new markets will depend strongly on its structure-related properties. The molar mass distribution, for example, partly governs its reactivity and physico-chemical properties.
The unique exception is the biorefinery operated by Borregaard in Norway. Here, 90% of the woody biomass input exits as marketable products. The refinery separates the biomass into cellulose specialty fibers (dissolving cellulose) and lignosulfonates. Part of the latter is then converted to vanillin, while the dissolved carbohydrates are fermented into 2nd-generation bioethanol.59 In this way, at least one high-value chemical is produced from lignin. Vanillin is the world's most used flavoring agent. Its production is about 16 ktpa (vanillin from vanilla beans represents only 1% of that60). Extensive reviews covering methods for obtaining and purifying vanillin were published previously.61–64 Besides vanillin, lignin oxidation with molecular oxygen produces syringaldehyde and various aromatic aldehydes.65 Syringaldehyde is a valuable starting material for the pharmaceutical industry, being a precursor for the synthesis of 3,4,5-trimetoxybenzaldehyde.66 Notably, the presently efficiency of catalytic oxidation lignin by molecular oxygen is very close to the selectivity of the oxidation by nitrobenzene.67,68 The latter is considered to be the theoretically highest possible yield of aromatic aldehydes from lignin.69–71 Interestingly, converting lignin to aromatics used to be more common in industry, though vanillin remains today the only commercial aromatic product of lignin. In 1982, Huibers and Parkhurst reported the highest yields for the conversion of kraft lignin into aromatics.72 In a two-step process, including hydrocracking and hydrodealkylation over a catalyst bed in hydrogen, 20% of phenol and 14% of benzene (based on lignin) were isolated.72 Considering that today's phenol market price is about 1220 € per ton and benzene 1050 € per ton (January 2014 prices), this could become a valuable process in a long- to medium-term application. The remaining 70% could still be burned to generate power, increasing the overall revenue. Currently, 95% of phenol production involves the partial oxidation of cumene (isopropylbenzene) via the Hock process. In the past two decades, much research was dedicated to synthesize phenol via the direct oxidation of benzene.73,74 This could avoid the co-production of acetone, offering the potential economy of operation without by-product. As phenol is more expensive than benzene, producing phenol from lignin can be even more attractive.
More and more lignin is available as technologies continuously improve. Hence more added value chemicals should be targeted within the lignin stream. Even if only 25% of lignin will be isolated from 600 Mtons of biomass (only half of the US prediction by 2060), it would mean that 150 Mtons of isolated lignin would be available in the U.S.75 The market price of technical lignin is about 110 € per ton.76,77 According to Holladay et al.,22 in a scenario where lignin is used as a fuel to generate power, the fuel credit value is €8.6bn.75,78 But, rather than replacing fossil fuels, lignin should be converted into high-value aromatics and fine-chemicals. This market is much more lucrative. The global production of benzene, toluene and xylene (BTX) in 2012 was 102 Mtpa.79 The market price of BTX was reported on average of 876 Euro per ton in 2010.80 This represents a value of €73bn.80 Replacing this type of market with lignin is economically much more interesting. These products could fed into conventional petrochemical processes, reducing the investment in new platform refineries (see Fig. 5).
 | ||
| Fig. 5 Current petrochemical products starting from benzene and phenol. | ||
In a scenario where lignin is converted only to simple aromatics, with a 20 wt% conversion, 372 Mtons of BTX are produced at a value of €19bn.22,81 But producing BTX selectively from lignin remains a challenge. Most processes deal with the conversion of a mixture of aromatic structures, increasing the difficulty of purification steps. Even if BTX would be produced in a mixture of other aromatics (80% of lignin), the benefit would be ca. €24bn, with the remainder converted to 290 Mtons of bio-ethanol.22,81 The mixture of compounds would be gasified to provide syngas alcohols with added value. Regrettably, lignin valorisation research has not improved since the 80's on a commercial scale. Much effort is now done on a labscale to understand and improve the different catalytic steps and overcome the challenge of pilot scale will need some extra time. The prize is tempting – compared with the €8.6bn for power generation, BTX production could double the added value of lignin income.
Using lignin as a low-cost carbon source can decrease the sensitivity of the raw material to petroleum cost. The goal of replacing steel panels with lightweight, yet strong, carbon fibre-reinforced plastics is to significantly reduce vehicle weight and improve fuel transportation economy. The carbon fibre production requires approx. 0.6 billion kg, composed of 90% lignin and 10% synthetic polymer with an overall weight yield of 45%. In a scenario where lignin is converted to 7.6 million tons of carbon fibres, considering that 45 kg of carbon fibres are needed per car, this would go into 15 million cars. The remaining lignin would be converted to syngas and alcohols, which would increase the ethanol production of 360 Mtons and the revenue increase of €12bn.81
In agrochemicals sector, lignin can also have an impact. Lignosulfonates are already used commercially in many applications such as in the cement, polymers, resins and foams sectors. They are excellent dispersing agents, binders and dust controlling properties. Will and Yokose estimated the world production value in 2005 between €360m and €400m.86 As mentioned above, the majority of the lignosulfonates comes from sulfite pulping and fibre industry.
One of the largest applications of lignosulfonates is in the concrete industry. About 700 ktpa of lignosulfonates are used only in this field.86 Their strong dispersing agents properties allows using less water to form workable mixtures.87 Thanks to lower water requirements, the resulting concrete has a higher density, better uniformity, higher compressive strength and better durability. Lignosulfonates are also used as set retarding agents in applications where concrete must remain fluid over extended periods of time. A key factor in cement deposition is controlling dust and surface stabilization.88 Lignosulfonates are typically sprayed on the roads to prevent dust formation. In the form of potassium lignosulfonates, they can be used in up till 30 wt% in slow-release fertilizers. This improves the chelating of a variety of metal ions in the soil as well as increases the solubility of nitrogen, phosphorus and potassium in the soil.72 Their binding properties increase hardness and avoid cracking and dust in storing, from animal feed pellets to fertilizers.89,90 In the past 30 years, numerous environmental studies were conducted to evaluate the toxicity risks and gas emission on the use of lignosulfonates. They concluded that lignosulfonates are non-toxic above the concentration level used, and that the surrounding vegetation is not affected by their application.91–93
Polyurethane is one of the most versatile polymers, with a wide range of products in diverse sectors, as a foam, elastomer, paints and adhesive. With its high insulation and mechanical properties, rigid polyurethane (RPU), with foams and elastomers are often used in the construction, automotive industry, freeze sectors, equipment manufacturing, nautical applications.94 The utilization of lignin in polyurethane synthesis often follows two global approaches: the direct utilization of lignin without any preliminary chemical modification (alone or in combination with other polyols)95,96 or by making the hydroxyl functions more readily available by chemical modification such as esterification and etherification reactions.97 As lignin contains both aliphatic and aromatic hydroxyl groups, it can potentially act as reactive sites for isocyanate groups (formation of urethane linkages). It can be directly incorporated into polyurethane formulations without any chemical transformation. Silva et al. outlined five synthetic routes for incorporating lignin into polyurethane materials (see Fig. 6).98
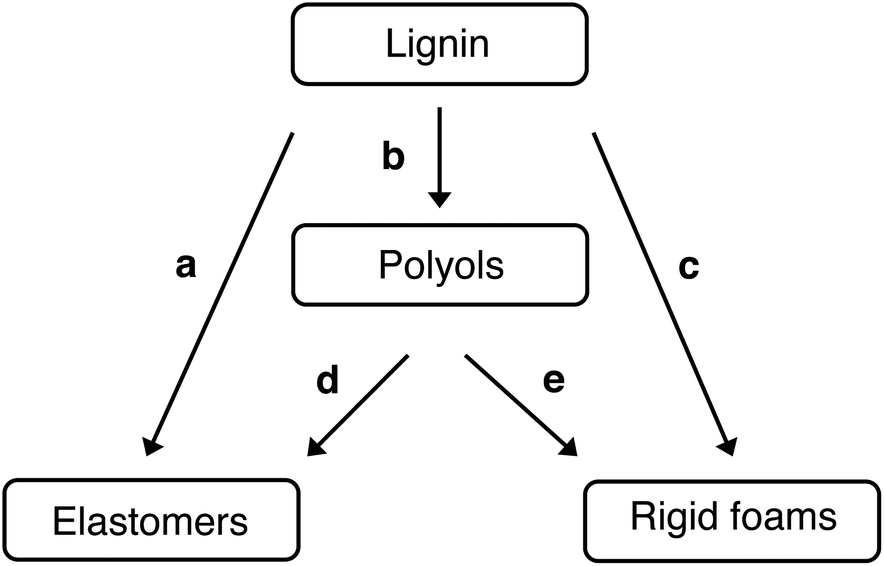 | ||
| Fig. 6 Synthetic routes for lignin incorporation in polyurethane materials: (a) direct use, (b) synthesis of liquid polyol, (c) synthesis of rigid polyurethane foams using lignin-based polyols, (e) synthesis of polyurethane elastomers using lignin-based polyols and (d) synthesis of rigid polyurethane foams using lignin as reactive filler.98 | ||
The drawback is a more rigid and brittle end product compared to pure polyurethane. To overcome this, oxypropylation is used. This reaction grafts the poly(propylene oxide) on the lignin, allowing the hydroxyl groups to be released on the outer shell of the polymer (particularly the phenolic groups entrapped inside the molecule).81 Consequently, the solid lignin becomes a liquid polyol with an optimal hydroxyl index for polyurethane foam formulations. Both approaches, using lignin directly or after chemical modification, formed materials with promising properties.98
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 blend of lignin–polyethylene oxide.101 The aromatic character of lignin makes it a good starting material for graphite electrodes, as shown by Popova et al.102 and more recently by the groups of McGuire and Gnedenkov.103,104 These can be used in both lithium batteries and fuel cells.
:1 blend of lignin–polyethylene oxide.101 The aromatic character of lignin makes it a good starting material for graphite electrodes, as shown by Popova et al.102 and more recently by the groups of McGuire and Gnedenkov.103,104 These can be used in both lithium batteries and fuel cells.The problem is that each of these limitations is being tackled by different groups or business units, that often compete rather than collaborate with each other. This may sound trivial, but the problems of lignin valorisation are too far apart to be solved by single groups or companies.113 Collaborative projects are essential.
Tackling the product application issues needs a more fundamental change. It requires a shift in the buyers' view on chemicals, because of the inherent variety in biomass feeds. Some lignin extracts will serve as feedstocks for traditional products which require high-purity components. But we foresee a major market segment for aromatic “cocktails”, provided that the variety in the feed would not hamper the performance. As an example, consider linear alkylated benzenes (LABs),114 which are used for making surfactants and ultimately cosmetics. Since the active ingredients in these are typically formulated using several components, we can envisage a lignin-based cocktail of LABs that would be applied “as is” for making surfactant mixtures. This would be the best long-term approach for using biomass-derived feeds, as they are and will remain varied by location and time.
Another major problem of lignin valorization is the hydrogen source. Converting lignin to valuable chemicals involves various steps, including hydrocracking, hydrodealkylation and hydrodeoxygenation. All these steps use large amount of hydrogen, for which fossil fuels will still remain the main source.
Conclusions
Lignin is nature's largest reserve of aromatics. Currently, with the exception of the vanillin process and the cement dispersion applications, it is burned as low-grade boiler fuel. Considering its complexity and richness of functional groups, simply burning lignin is a waste. There are many possible alternatives for converting it instead into aromatics, agrochemicals, polymers and high-performance materials such as carbon fibres. However, all these processes depend on improvements and innovations in the fields of catalysis and product separation. The catalytic steps that need improvement include dealkylations and hydrodeoxygenation. Moreover, new types of catalysts are needed,115 that are compatible with biomass feedstocks. Most importantly, realizing the full industrial potential of lignin requires large-scale and intensive collaboration between scientists and engineers in the fields of analytics, catalysis, organic chemistry, and chemical engineering and processing.Acknowledgements
This research was performed within the framework of the CatchBio program. The authors gratefully acknowledge the support of the Smart Mix Program of the Netherlands Ministry of Economic Affairs and the Netherlands Ministry of Education, Culture and Science.Notes and references
- I. V. Babich and J. A. Moulijn, Fuel, 2003, 82, 607–631 CrossRef CAS.
- M. S. Rana, V. Sàmano, J. Ancheyta and J. A. I. Diaz, Fuel, 2007, 86, 1216–1231 CrossRef CAS PubMed.
- M. K. Hubbert, Nuclear Energy and the Fossil Fuels, American Petroleum Institute, Drilling and Production Practice, in Proceedings of Spring Meeting, San Antonio, 1956, pp. 7–25 Search PubMed.
- S. Sorrell, J. Speirs, R. Bentley, A. Brandt and R. Miller, Energy Policy, 2010, 38, 5290–5295 CrossRef PubMed.
- K. Aleklett, M. Höök, K. Jakobsson, M. Lardelli, S. Snowden and B. Söderbergh, Energy Policy, 2010, 38, 1398–1414 CrossRef PubMed.
- P. de Almeida and P. D. Silva, Energy Policy, 2009, 37, 1267–1276 CrossRef PubMed.
- G. Maggio and G. Cacciola, Fuel, 2012, 98, 111–123 CrossRef CAS PubMed.
- A. A. Kiss, F. Omota, A. C. Dimian and G. Rothenberg, Top. Catal., 2006, 40, 141–150 CrossRef CAS PubMed.
- Y. Sun and J. Cheng, Bioresour. Technol., 2002, 83, 1–11 CrossRef CAS.
- World Energy Outlook 2010–12, IEA, International Energy Agency, Paris, 2010 Search PubMed.
- C. Zou, D. Dong, S. Wang, J. Li, X. Li, Y. Wang, D. Li and K. Cheng, Pet. Explor. Dev., 2010, 37, 641–653 CrossRef CAS.
- F. Carvalheiro, L. C. Duarte and F. M. Gìrio, J. Sci. Ind. Res., 2008, 67, 849–864 CAS.
- S. Fernando, S. Adhikari, C. Chandrapal and N. Murali, Energy Fuels, 2006, 20, 1727–1737 CrossRef CAS.
- K. J. Kudakasseril, N. G. Raveendran, A. Hussain and R. G. S. Vijaya, Renewable Sustainable Energy Rev., 2013, 25, 205–219 CrossRef PubMed.
- M. Foston and A. J. Ragauskas, Ind. Biotechnol., 2012, 8, 191–208 CrossRef CAS.
- P. N. R. Vennestrøm, C. M. Osmundsen, C. H. Christensen and E. Taarning, Angew. Chem., Int. Ed., 2011, 50, 10502–10509 CrossRef PubMed.
- A. Jess and P. Wasserscheid, Chemical technology - An Integrated Textbook, Wiley-VCH, Weinheim, 2013 Search PubMed.
- T. Wiesenthal, A. Mourelatou, J.-E. Peterson and P. Taylor, How much bioenergy can Europe produce without harming the environment?, European Environment Agency, Copenhagen, 2006 Search PubMed.
- R. D. Perlack, et al., Biomass as Feedstock for a Bioenergy and Bioproducts Industry: The Technical Feasibility of a Billion-Ton Annual Supply, U.S. Department of Energy and U.S. Department of Agriculture, 2005 Search PubMed.
- A. J. Ragauskas, C. K. Williams, B. H. Davison, G. Britovsek, J. Cairney, C. A. Eckert, W. J. Frederick, J. P. Hallett, D. J. Leak, C. L. Liotta, J. R. Mielenz, R. Murphy, R. Templer and T. Tschaplinski, Science, 2006, 311, 484–489 CrossRef CAS PubMed.
- N. R. Shiju, D. R. Brown, K. Wilson and G. Rothenberg, Top. Catal., 2010, 53, 1217–1223 CrossRef CAS.
- J. E. Holladay, J. F. White, J. J. Bozell and D. Johnson, Top Value-Added Chemicals from Biomass: Volume II—Results of Screening for Potential Candidates from Biorefinery Lignin, Pacific Northwest National Laboratory, 2007 Search PubMed.
- P. Demma Carà, M. Pagliaro, A. Elmekawy, D. R. Brown, P. Verschuren, N. R. Shiju and G. Rothenberg, Catal. Sci. Technol., 2013, 3, 2057–2061 Search PubMed.
- M. J. Louwerse and G. Rothenberg, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 035108 CrossRef.
- Y. Zheng, E. K. Yanful and A. S. Bassi, Crit. Rev. Biotechnol., 2005, 25, 243–250 CrossRef CAS PubMed.
- C. Witthayaprapakorn, K. Thananukul, N. Suttenun, P. Punyamoonwongsa and R. Molloy, Adv. Mater. Res., 2013, 664, 702–706 CrossRef CAS , 706 pp.
- C. Xiang, Y. L. Joo and M. W. Frey, J. Biobased Mater. Bioenergy, 2009, 3, 147–155 CrossRef CAS PubMed.
- S. Pirani, H. M. N. Abushammala and R. Hashaikeh, J. Appl. Polym. Sci., 2013, 130, 3345–3354 CrossRef CAS.
- S. C. Cruz, G. Rothenberg, J. A. Westerhuis and A. K. Smilde, Chemom. Intell. Lab. Syst., 2008, 91, 101–109 CrossRef CAS PubMed.
- Y. Poirier, C. Nawrath and C. Somerville, Nat. Biotechnol., 1995, 13, 142–150 CrossRef CAS PubMed.
- N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., Int. Ed., 2011, 50, 9615–9619 CrossRef CAS PubMed.
- A. S. V. De Sousa Dias, G. J. M. Gruter and R.-J. Van Putten, Process for the conversion of a carbohydrate-containing feedstock, WO 2012/091770 A1, 2010.
- E.-J. Ras, B. McKay and G. Rothenberg, Top. Catal., 2010, 53, 1202–1208 CrossRef CAS.
- M. Zhang and Y. Yu, Ind. Eng. Chem. Res., 2013, 52, 9505–9514 CrossRef CAS.
- Z. W. Srokol and G. Rothenberg, Top. Catal., 2010, 53, 1258–1263 CrossRef CAS.
- J. K. Bourne and R. Clark, National Geographic, 2007, 212, 38–59 Search PubMed.
- J.-K. Weng, X. Li, N. D. Bonawitz and C. Chapple, Curr. Opin. Biotechnol., 2008, 19, 166–172 CrossRef CAS PubMed.
- S. K. Ritter, Chem. Eng. News, 2008, 46, 15 Search PubMed.
- F. S. Chakar and A. J. Ragauskas, Ind. Crops Prod., 2004, 20, 131–141 CrossRef CAS PubMed.
- R. J. Gosselink, PhD thesis, Wageningen University, 2011.
- J. H. Lora, C. F. Wu, E. K. Pye and J. J. Balatinecz, Characteristics and Potential Applications of Lignin Produced by an Organosolv Pulping Process, American Chemical Society, 1989, vol. 397, pp. 312–323 Search PubMed.
- G. Brunow, Methods to Reveal the Structure of Lignin, in Biopolymers, Volume 1: Lignin, Humic Substances and Coal, ed. M. Hofrichter and A. Steinbüchel, Wiley-VCH, Weinheim, 2001, vol. 1, p. 106 Search PubMed.
- J. Zakzeski, P. C. A. Bruijnincx, A. L. Jongerius and B. M. Weckhuysen, Chem. Rev., 2010, 110, 3552–3599 CrossRef CAS PubMed.
- R. Vanholme, B. Demedts, K. Morreel, J. Ralph and W. Boerjan, Plant Physiol., 2010, 153, 895–905 CrossRef CAS PubMed.
- J. Gierer, Wood Sci. Technol., 1980, 14, 241–266 CrossRef CAS.
- E. Adler, Wood Sci. Technol., 1977, 11, 169–218 CrossRef CAS.
- C. L. Chen, Int. Fiber Sci. Technol. Ser., 1991, 11, 183–261 CAS.
- R. Ede and I. Kilpeläinen, Res. Chem. Intermed., 1995, 21, 313–328 CrossRef CAS.
- P. Karhunen, P. Rummakko, J. Sipilä, G. Brunow and I. Kilpeläinen, Tetrahedron Lett., 1995, 36, 169–170 CrossRef CAS.
- J. Lora, B. M. Naceur and A. Gandini, Industrial commercial lignins: sources, properties and applications, Elsevier, Amsterdam, 2008, p. 225 Search PubMed.
- A. Abächerli and F. Doppenberg, Treatment process of alkali solutions containing aromatic polymers, CZ9903156, 2000.
- F. Öhman, H. Theliander, P. Tomani and P. Axegard, Method for separating lignin from black liquor, US 2008/0047674, 2008.
- A. G. Maldonado and G. Rothenberg, Chem. Eng. Prog., 2009, 105, 26–32 CAS.
- A. V. Gaikwad, P. Verschuren, T. van der Loop, G. Rothenberg and E. Eiser, Soft Matter, 2009, 5, 1994–1999 RSC.
- X. Pan and J. N. Saddler, Biotechnol. Biofuels, 2013, 6, 12 CrossRef CAS PubMed.
- A. Higson and C. Smith, Renewable Chemicals Factsheet: Lignin, NNFCC, 2011.
- P. Azadi, O. R. Inderwildi, R. Farnood and D. A. King, Renewable Sustainable Energy Rev., 2013, 21, 506–523 CrossRef CAS PubMed.
- P. Gallezot, ChemSusChem, 2008, 1, 734–737 CrossRef CAS PubMed.
- Borregaard accessed on January 2014, http://www.borregaard.com/content/view/full/10231.
- R. T. Winter, H. L. van Beek and M. W. Fraaije, J. Chem. Educ., 2011, 89, 258–261 CrossRef.
- K. Anuradha, B. N. Shyamala and M. M. Naidu, Crit. Rev. Food Sci. Nutr., 2013, 53, 1250–1276 CrossRef CAS PubMed.
- F. G. Calvo-Flores and J. A. Dobado, ChemSusChem, 2010, 3, 1227–1235 CrossRef CAS PubMed.
- E. A. B. da Silva, M. Zabkova, J. D. Araujo, C. A. Cateto, M. F. Barreiro, M. N. Belgacem and A. E. Rodriques, Chem. Eng. Res. Des., 2009, 87, 1276–1292 CrossRef PubMed.
- W. B. Huang, C. Y. Du, J. A. Jiang and Y. F. Ji, Res. Chem. Intermed., 2013, 39, 2849–2856 CrossRef CAS PubMed.
- P. C. R. Pinto, E. A. B. da Silva and A. E. Rodriques, Lignin as source of fine chemicals: vanillin and syringaldehyde, ed. C. Baskar, S. Baskar and R. S. Dhillon, Springer, Heidelberg, 2012, pp. 381–420 Search PubMed.
- Y. V. Erofeev, V. L. Afanaseva and R. G. Glushkov, Pharm. Chem. J., 1990, 24, 501–510 CrossRef.
- V. E. Tarabanko, Y. V. Chelbina, A. V. Kudryashev and N. V. Tarabanko, Sep. Sci. Technol., 2013, 48, 127–132 CrossRef CAS.
- V. E. Tarabanko, Y. V. Chelbina, V. A. Sokolenko and N. V. Tarabanko, Solvent Extr. Ion Exch., 2007, 25, 99–107 CrossRef CAS.
- F. Brauns and D. Brauns, The chemistry of lignin, Academic Press, New York, 1952 Search PubMed.
- T. Elder, ACS Symp. Ser., 1989, 397, 262–271 CrossRef CAS PubMed.
- H. E. Schoemaker, Recl. Trav. Chim. Pays-Bas, 1990, 109, 255–272 CrossRef CAS.
- D. T. A. Huibers and H. J. Parkhurst, Jr., Lignin hydrocracking process to produce phenol and benzene, US 4420644 A, 1982.
- D. Bianchi, R. Bortolo, R. Tassinari, M. Ricci and R. Vignola, Angew. Chem., Int. Ed., 2000, 39, 4321–4323 CrossRef CAS.
- W. Laufer and W. F. Hoelderich, Chem. Commun., 2002, 0, 1684–1685 RSC.
- T. Werpy, G. Petersen, A. Aden, J. Bozell, J. Holladay, J. White, A. Manheim, D. Eliot, L. Lasure and S. Jones, Top Value Added Chemicals from Biomass. Volume 1-Results of Screening for Potential Candidates from Sugars and Synthesis Gas, DTIC Document, 2004 Search PubMed.
- M. R. Olsson, E. Axelsson and T. Berntsson, Nord. Pulp Pap. Res. J., 2006, 21, 476–484 CAS.
- T. Voitl and P. R. v. Rohr, Ind. Eng. Chem. Res., 2009, 49, 520–525 CrossRef.
- A. Popov, E. Kondratieva, J. M. Goupil, L. Mariey, P. Bazin, J.-P. Gilson, A. Travert and F. Maugé, J. Phys. Chem. A, 2010, 114, 15661–15670 CAS.
- J. van Haveren, E. L. Scott and J. Sanders, Biofuels, Bioprod. Biorefin., 2008, 2, 41–57 CrossRef CAS.
- Chemical Economics Handbook Product review, SRI International, http://www.ihs.com, 2011.
- J. P. S. Aniceto, I. Portugal and C. M. Silva, ChemSusChem, 2012, 5, 1358–1368 CrossRef CAS PubMed.
- T. Saito, J. H. Perkins, F. Vautard, H. M. Meyer, J. M. Messman, B. Tolnai and A. K. Naskar, ChemSusChem, 2014, 7, 221–228 CrossRef CAS PubMed.
- J. F. Kadla, S. Kubo, R. A. Venditti, R. D. Gilbert, A. L. Compere and W. Griffith, Carbon, 2002, 40, 2913–2920 CrossRef CAS.
- L. Duran Pachon, I. Yosef, T. Z. Markus, R. Naaman, D. Avnir and G. Rothenberg, Nat. Chem., 2009, 1, 160–164 CrossRef CAS PubMed.
- C. Eberle, D. C. Webb, T. Albers and C. Chen, Commercialization of New Carbon Fiber Materials Based on Sustainable Resources for Energy Applications, Oak Ridge National Laboratory, 2013 Search PubMed.
- T. Stern and P. Schwarzbauer, For. Prod. J., 2008, 58, 81–86 CAS.
- N. P. Mailvaganam and M. Rixom, Chemical admixtures for concrete, Taylor & Francis, 2002 Search PubMed.
- F. M. Ernsberger and W. G. France, Ind. Eng. Chem. Res., 1945, 37, 598–600 CrossRef CAS.
- N. Acar, E. Moran, W. Revington and S. Bilgili, Poult. Sci., 1991, 70, 1339–1344 CrossRef.
- W. J. Detroit, Lignosulfonate treated fertilizer particles, US 5041153 A, 1989.
- J. W. Adams, Environmental Effects of Applying Lignosulfonate to Roads, Daishowa chemicals Incorporated, 1988 Search PubMed.
- J. B. Nelson and R. A. Northey, J. Environ. Manage., 2004, 73, 333–338 CrossRef PubMed.
- M. A. Stapanian and D. W. Shea, Int. J. Environ. Stud., 1986, 27, 45–56 CrossRef CAS.
- J. S. Amaral, M. Sepùlveda, C. A. Cateto, I. P. Fernandes, A. r. E. Rodrigues, M. N. Belgacem and M. F. Barreiro, Polym. Degrad. Stab., 2012, 97, 2069–2076 CrossRef CAS PubMed.
- C. A. Cateto, M. F. Barreiro, A. E. Rodrigues, M. C. Brochier-Salon, W. Thielemans and M. N. Belgacem, J. Appl. Polym. Sci., 2008, 109, 3008–3017 CrossRef CAS.
- M. Vanderlaan and R. Thring, Biomass Bioenergy, 1998, 14, 525–531 CrossRef CAS.
- H. Nadji, C. Bruzzèe, M. N. Belgacem, A. Benaboura and A. Gandini, Macromol. Mater. Eng., 2005, 290, 1009–1016 CrossRef CAS.
- E. Silva, M. Zabkova, J. Araùjo, C. Cateto, M. Barreiro, M. Belgacem and A. Rodrigues, Chem. Eng. Res. Des., 2009, 87, 1276–1292 CrossRef PubMed.
- R. B. Lima, R. Raza, H. Qin, J. Li, M. E. Lindstroem and B. Zhu, RSC Adv., 2013, 3, 5083–5089 RSC.
- H. H. Weetall, B. D. Forsyth and W. Hertl, Biotechnol. Bioeng., 1985, 27, 972–979 CrossRef CAS PubMed.
- S.-X. Wang, L. Yang, L. P. Stubbs, X. Li and C. He, ACS Appl. Mater. Interfaces, 2013, 5, 12275–12282 CAS.
- O. V. Popova, S. S. Popova and L. N. Ol'shanskaya, Russ. J. Appl. Chem., 2008, 81, 786–791 CrossRef CAS.
- W. E. Tenhaeff, O. Rios, K. More and M. A. McGuire, Adv. Funct. Mater., 2014, 24, 86–94 CrossRef CAS.
- S. V. Gnedenkov, D. P. Opra, S. L. Sinebryukhov, A. K. Tsvetnikov, A. Y. Ustinov and V. I. Sergienko, J. Solid State Electrochem., 2013, 17, 2611–2621 CrossRef CAS.
- E. A. Capanema, M. Y. Balakshin, C.-L. Chen, J. S. Gratzl and H. Gracz, Holzforschung, 2001, 55, 302–308 CAS.
- J. C. del Rio, J. Rencoret, P. Prinsen, A. T. Martinez, J. Ralph and A. Gutierrez, J. Agric. Food Chem., 2012, 60, 5922–5935 CrossRef CAS PubMed.
- S. Brudin, J. Berwick, M. Duffin and P. Schoenmakers, J. Chromatogr. A, 2008, 1201, 196–201 CrossRef CAS PubMed.
- S. Brudin and P. Schoenmakers, J. Sep. Sci., 2010, 33, 439–452 CrossRef CAS PubMed.
- H.-J. Huang, S. Ramaswamy, U. W. Tschirner and B. V. Ramarao, Sep. Purif. Technol., 2008, 62, 1–21 CrossRef CAS PubMed.
- A. C. Marr and S. Liu, Trends Biotechnol., 2011, 29, 199–204 CrossRef CAS PubMed.
- C. E. Wyman and B. J. Goodman, Appl. Biochem. Biotechnol., 1993, 39–40, 41–59 CrossRef CAS.
- P. N. R. Vennestroem, C. H. Christensen, S. Pedersen, J.-D. Grunwaldt and J. M. Woodley, ChemCatChem, 2010, 2, 249–258 CrossRef CAS.
- J. H. Clark, F. E. I. Deswarte and T. J. Farmer, Biofuels, Bioprod. Biorefin., 2009, 3, 72–90 CrossRef CAS.
- C. Perego and P. Ingallina, Catal. Today, 2002, 73, 3–22 CrossRef CAS.
- N. R. Shiju, A. H. Alberts, S. Khalid, D. R. Brown and G. Rothenberg, Angew. Chem., Int. Ed., 2011, 50, 9615–9619 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |