
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiamond functionalization with light-harvesting molecular wires: improved surface coverage by optimized Suzuki cross-coupling conditions†
W. S.
Yeap‡
*a,
D.
Bevk‡
ab,
X.
Liu‡
c,
H.
Krysova‡
d,
A.
Pasquarelli‡
e,
D.
Vanderzande‡
ab,
L.
Lutsen‡
ab,
L.
Kavan‡
d,
M.
Fahlman‡
c,
W.
Maes‡
ab and
K.
Haenen‡
*ab
aHasselt University, Institute for Materials Research (IMO), B-3590 Diepenbeek, Belgium. E-mail: wengsiang.yeap@uhasselt.be; Fax: +32 11 26 8899; Tel: +32 11 26 8826
bIMEC vzw, IMOMEC, B-3590 Diepenbeek, Belgium. E-mail: ken.haenen@uhasselt.be
cLinköping University, Department of Physics, Chemistry and Biology, S-58183 Linköping, Sweden
dAcademy of Sciences of The Czech Republic, J. Heyrovský Institute of Physical Chemistry, v.v.i., Dolejškova 3, CZ-182 23 Prague 8, Czech Republic
eUlm University, Institute of Electron Devices and Circuits, 89069 Ulm, Germany
First published on 29th August 2014
Abstract
Donor–acceptor type light-harvesting molecular wires are covalently attached to a boron-doped diamond surface via a combination of diazonium electrografting and Suzuki cross-coupling. For the Suzuki reaction, various catalytic systems are compared with respect to their imposed surface coverage. Combining 2-dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos) and Pd(0), the diamond coverage improves considerably (by 98%) as compared to the standard tetrakis(triphenylphosphine)palladium(0) (Pd(PPh3)4) catalyst. As the energy levels between the molecular chromophores and the diamond film align well, the sophisticated functionalized diamond surfaces present a first step towards the development of fully carbon-based devices for light to electricity conversion.
Introduction
In recent years, boron-doped diamond (BDD) has become increasingly important within science and technology.1 By adding boron, diamond conductivity can be tuned, with values ranging from 1 × 10−9 to 100 Ω−1 cm−1, the latter resulting in metallic behavior.2 Boron-doped nanocrystalline diamond (B:NCD) prepared with a thickness of ∼150 nm is highly transparent (with a transmission of 70–80%).3 Despite its extraordinary physical properties and numerous applications (chemical and biosensors, electrosynthesis etc.),4 earlier works focused mostly on tailoring the diamond's surface properties. Photochemical modification,5 plasma treatment,6 and diazonium coupling7 afford a diversity of diamond surface modification schemes, allowing the attachment of various molecular structures. The next challenge, however, is to develop a versatile chemistry that can impart useful sensory, optoelectronic and/or photophysical properties to the B:NCD material.Next to several click chemistry protocols,8 the Suzuki–Miyaura reaction represents one of the most versatile and powerful carbon–carbon (most often aryl–aryl) bond forming procedures in modern synthetic chemistry.9 This Pd-catalyzed cross-coupling reaction has been applied for the preparation of a huge variety of conjugated aromatic compounds, for example to be used for organic light-emitting diodes (OLEDs),10 polymer LEDs,11 organic photovoltaics,12 and nonlinear optical materials.13 The Suzuki reaction can also be used for (uninterrupted) molecular conjugation of solid surfaces to a large class of organic molecules.14 This is particularly useful if one wishes to create donor–acceptor type molecular wires on semiconducting substrates for photocurrent/photovoltage generation.15 However, to the best of our knowledge, there are only three reports so far which address Suzuki cross-coupling reactions on diamond surfaces.16–18 Zhong et al. reported a facile method for the Suzuki coupling of oligothiophene-fullerene and 2-(dicyanovinyl)-5-iodobithiophene molecular wires onto diamond. They showed that this method opens up possibilities for the application of diamond in molecular electronics and photovoltaics. Nonetheless, the reported surface coverage of 2-(dicyanovinyl)-5-iodobithiophene was as low as 0.41 ‘monolayer’ (ML) (relative to a high quality reference monolayer of dodecanethiol on Au, as determined by X-ray photoelectron spectroscopy, XPS).17
In this manuscript, we report on the successful decoration of B:NCD with two molecular wire systems, (Z)-2-{5-[(5′-bromo-[2,2′-bithiophen]-5-yl)methylene]-4-oxo-2-thioxothiazolidin-3-yl}acetic acid (Br-BT-Rho) and (E)-2-{4-[2-(6-bromo-4,4-diethyl-4H-cyclopenta[1,2-b:5,4-b′]dithiophen-2-yl)vinyl]-3-cyano-5,5-dimethylfuran-2(5H)-ylidene}malononitrile (Br-CPDT-Fur) (Scheme 1), via optimized Suzuki cross-coupling reactions. Both molecular wires possess a bromoaryl moiety to enable reaction with an arylboronic ester functionalized B:NCD surface. The most effective catalytic system was pursued to increase the coupling efficiency and hence the surface coverage. Surface functionalization as well as coverage were determined by XPS. The CPDT-Fur molecular wire has suitable properties to be used effectively in future molecular photovoltaic systems, i.e. high absorbance, wide absorption and good charge transfer abilities through the π-system.19 Furthermore, the highest occupied molecular orbital (HOMO) of the dye seems to match well with the valence band of the B:NCD film, as evaluated by ultraviolet photoelectron spectroscopy (UPS), allowing hole injection from the dye to diamond.
 | ||
| Scheme 1 Diazonium electrografting and Suzuki cross-coupling towards effective B:NCD functionalization. | ||
Results and discussion
As shown in Scheme 1, the anchoring of the molecular wires onto the diamond surface begins with the functionalization of hydrogen-terminated B:NCD (see Experimental) with arylboronic ester moieties through diazonium electrografting.16 Two light-harvesting molecular wires with a donor–acceptor structure (to increase light-harvesting and enhance charge separation), Br-BT-Rho and Br-CPDT-Fur (Scheme 1), were pre-synthesized in solution before undergoing Suzuki coupling to the B:NCD surfaces. Both molecular wires contain an electron-rich (bridged) bithiophene ‘donor’ part and an electron-deficient subunit and they carry a reactive arylbromide group on the donor part. As the B:NCD film is finally intended to be used as the hole conductor in photovoltaic devices, the donor part of the chromophores has to be attached to the diamond surface. The Br-BT-Rho chromophore was synthesized by reacting commercially available 5-bromo-5′-formyl-2,2′-bithiophene with rhodanine-3-acetic acid in a standard condensation reaction. On the other hand, the Br-CPDT-Fur molecule was synthesized according to the procedure outlined in Scheme 2. | ||
| Scheme 2 Synthetic protocol towards the Br-CPDT-Fur molecular wire. | ||
4H-Cyclopenta[1,2-b:5,4-b′]dithiophenes (CPDTs) have been proven to be attractive electron donating building blocks for applications in organic electronics due to the planarity of the bridged bithiophene system and the excellent charge transfer capabilities through the molecule.19 For the purpose of B:NCD functionalization, a novel CPDT chromophore has been developed, employing a furan group as the acceptor part attached to the CPDT core via a vinyl bridge. The synthesis of the CPDT core was done according to two different previously published procedures.19a,20 CPDT formylation was performed using a standard Vilsmeier–Haack procedure in 1,2-dichloroethane at 0 °C. This reaction yielded exclusively monoformylated CPDT derivative 2, which was used in the next step without prior purification. Treatment of CPDT-CHO2 with N-bromosuccinimide (NBS) at room temperature furnished Br-CPDT-CHO derivative 3 in a smooth manner. To attach the furan acceptor, a Knoevenagel condensation reaction was performed in the presence of a catalytic amount of piperidine. The Br-CPDT-Fur dye precipitated from the hot reaction mixture and was recrystallized from the corresponding solvent and used as such. UV-Vis analysis of the dye showed a high extinction coefficient (ε = 73100 L mol−1 cm−1 at λ = 582 nm), thus proving suitability of the molecule for the envisaged application. The frontier orbital energy levels, as determined from the oxidation and reduction onsets derived from cyclic voltammetry (CV), are EHOMO = −5.70 eV and ELUMO = −4.03 eV.
Successful electrografting of the arylboronic ester groups on the B:NCD surface by the diazonium approach was confirmed by CV (Fig. 1). The first CV cycle shows two reduction peaks. The broader peak at −0.6 V (vs. Ag/AgCl) can be assigned to the formation of arylboronic ester radicals. During the second cycle, this reduction wave disappears and the cyclic voltammogram only exhibits a very small reduction current, which suggests passivation of the diamond surface by the grafted layer, hence blocking the access of the diazonium cations to the surface. A smaller reduction peak appearing at −0.1 V (vs. Ag/AgCl) was previously noticed as well for electrografting of 4-nitrophenyl- and 4-carboxyphenyl-diazonium cations, but its origin is not yet clearly understood.7
 | ||
| Fig. 1 In situ electrochemical generation of boronic ester derivatized phenyldiazonium cations, showing five CV reduction cycles (first cycle, black solid line; second cycle, blue dashed line; fifth cycle, red dotted line) with applied voltage between +0.5 and −0.8 V (scan rate 100 mV s−1). | ||
XPS spectra of the hydrogenated and arylboronic ester functionalized B:NCD films are shown in Fig. 2. As can be seen clearly from the high-resolution XPS spectra, the B 1s peak of the dopant boron atoms in B:NCD is found at ∼186 eV. After electrografting of the arylboronic ester groups on the B:NCD surface, an additional intense signal appears at ∼191 eV. This peak is assigned to the B 1s core level in boron atoms from the grafted arylboronic ester layer.16 High-resolution XPS clearly shows the presence of two chemically distinct boron species.
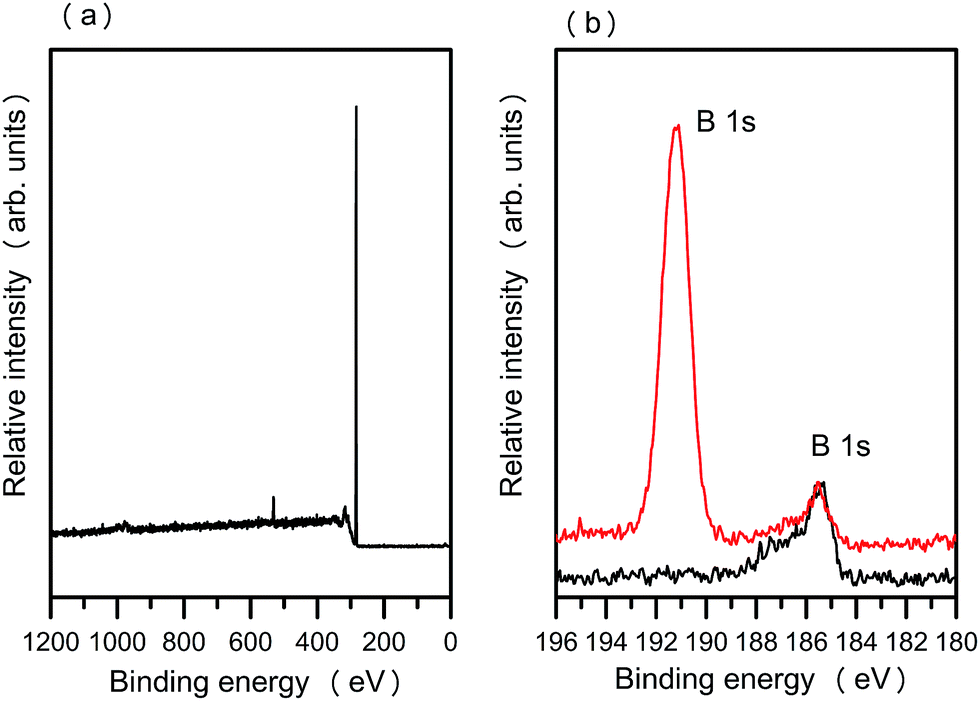 | ||
| Fig. 2 (a) XPS survey spectrum of hydrogenated B:NCD. (b) High-resolution XPS spectra showing the B 1s peaks for hydrogenated (black line) and arylboronic ester grafted B:NCD (red line), respectively. | ||
The arylboronic ester surface provides an excellent solid phase platform for further C–C extension via Suzuki cross-coupling. As such, an ‘all-carbon’ molecular photovoltaic system could be realized. However, a diamond surface with high chromophore loading is required for this purpose and this high coverage is not trivially obtained. To tackle this, a careful search for the optimum Suzuki conditions on arylboronic ester functionalized B:NCD was conducted. Tables 1 and 2 gather some of the different catalytic systems (changing the Pd catalyst, ligand, base and/or solvent) investigated for the two different chromophores (or their precursors). It should be mentioned at this point that the ‘opposite’ strategy, coupling 5-(5-formyl-2-thienyl)-2-thiopheneboronic acid ((HO)2B-BT-CHO) to a bromophenyl decorated B:NCD surface (obtained via spontaneous grafting of 4-bromobenzenediazonium tetrafluoroborate), was employed as well, but the obtained results were inferior to the procedure outlined here.
| Entry | Catalyst/liganda | Base | Solvent | Surface coverageb | |
|---|---|---|---|---|---|
| Br-BT-CHO c | Br-BT-Rho | ||||
| a General conditions: 80 °C, 18 h, 6 mL solvent, 100 μmol Ar–Br. b The XPS S 2p intensity was used to estimate the surface coverage. c Precursor used as a model compound. | |||||
| 1 | Pd(PPh3)4 (10 mol%) | NaOAc (1 equiv.) | THF | 0.05 | 0.01 |
| 2 | Pd(PPh3)4 (10 mol%) | Na2CO3 (4 equiv.) | THF–H2O (5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
0.02 | 0.005 |
| 3 | Pd(OAc)2 (5 mol%)–P(o-tol)3 (10 mol%) | K3PO4 (5 equiv.) | THF | 0.03 | — |
| 4 | Pd(OAc)2 (5 mol%)–P(o-tol)3 (10 mol%) | CsOAc (1 equiv.) | THF | 0.05 | — |
| 5 | Pd(OAc)2 (5 mol%)–P(o-tol)3 (10 mol%) | CsF (1 equiv.) | THF | Not successful | — |
| 6 | Pd(OAc)2 (5 mol%)–P(o-tol)3 (10 mol%) | NaOAc (1 equiv.) | THF | 0.24 | 0.08 |
| 7 | Pd(OAc)2 (3 mol%)–SPhos (3 mol%) | K3PO4 (5 equiv.) | Dioxane–H2O (5:1) |
0.43 | — |
| 8 | Pd(OAc)2 (3 mol%)–SPhos (3 mol%) | CsOAc (2 equiv.) | THF–MeOH (5:1) |
0.36 | 0.58 |
| Entry | Catalyst/liganda | Base | Solvent | Surface coverageb | |
|---|---|---|---|---|---|
| Br-CPDT-CHO c | Br-CPDT-Fur | ||||
| a General conditions: 80 °C, 18 h, 6 mL solvent, 100 μmol Ar–Br. b The XPS S 2p intensity was used to estimate the surface coverage. c Precursor used as model compound. | |||||
| 1 | Pd(PPh3)4 (10 mol%) | NaOAc (1 equiv.) | THF | 0.04 | 0.02 |
| 2 | Pd(OAc)2 (5 mol%)–P(o-tol)3 (10 mol%) | NaOAc (1 equiv.) | THF | — | 0.06 |
| 3 | Pd(OAc)2 (5 mol%)–P(o-tol)3 (10 mol%) | CsF (2 equiv.) | THF–MeOH (5:1) |
0.10 | — |
| 4 | Pd(OAc)2 (3 mol%)–SPhos (3 mol%) | K3PO4 (5 equiv.) | Dioxane–H2O (5:1) |
0.55 | — |
| 5 | Pd(OAc)2 (3 mol%)–SPhos (3 mol%) | CsF (2 equiv.) | THF–MeOH (5:1) |
0.06 | — |
| 6 | Pd(OAc)2 (3 mol%)–SPhos (3 mol%) | CsOAc (2 equiv.) | THF–MeOH (5:1) |
0.60 | 0.56 |
Successful coupling was confirmed by monitoring the S and N markers present in the dye molecules by XPS (Fig. 3). To determine the surface coverage, the S 2p peak intensities were calibrated against the XPS 2p intensity of a dodecanethiol self-assembled monolayer (SAM) on Au,17 where the coverage of the latter was assumed to be 1 ML (see Experimental). The coverages for both molecular wires as determined by this method are listed in Tables 1 and 2. When the standard catalytic system outlined in Entry 1 (Pd(PPh3)4, NaOAc, THF) was used, the obtained surface loadings (0.01 and 0.02 ML, respectively) were relatively low as compared to the results obtained by Zhong et al.17 There are three possible reasons for the low surface coverage. First, the relative reactivity for the Suzuki cross-coupling is known to be R–I (Zhong and co-workers used an iodinated bithiophene) > R–Br ≫ R–Cl.21 Secondly, Pd(PPh3)4 is known to be easily degrading, even in the presence of the smallest amount of oxygen, thus reducing the coupling efficiency.22 Thirdly, for sterically demanding molecules as the two molecular wires used here, catalytic systems with electron-rich ligands are often more appropriate.23 A slightly better coverage was observed when an in situ generated Pd catalyst, via combination of tri(o-tolyl)phosphine (P(o-tol)3) and palladium(II) acetate (Pd(OAc)2), was used (Entry 6, Table 1; Entry 2, Table 2). The more electron-donating tri(o-tolyl)phosphine ligand enhances the oxidative insertion of Pd into the Ar–Br bond.23 Moreover, the steric congestion of the ligand can also facilitate dissociation of the ligand from the Pd complex.
 | ||
| Fig. 3 XPS spectra showing the N 1s and S 2p peaks for the Br-CPDT-Fur (a and b) (Table 2, Entry 6) and Br-BT-Rho (c and d) (Table 1, Entry 8) based molecular wires (Pd(OAc)2/SPhos catalytic system). | ||
Over the past 20 years, the Buchwald group has made important contributions to the design of sterically hindered electron-rich biarylmonodentate phosphine ligands such as SPhos and XPhos (2-dicyclohexylphosphino-2′,4′,6′-triisopropylbiphenyl) which, when combined with catalytic amounts of Pd(0), enable to prepare extremely hindered biaryls via Suzuki–Miyaura cross-coupling reactions in an efficient manner.24 Recently, Zhou et al. successfully applied these conditions towards extension of the CPDT chromophore.25 Triggered by the effectiveness of the method and the similar CPDT building blocks used, we applied this catalytic system to the Suzuki cross-coupling on the B:NCD surface. Zhou and co-workers applied a dioxane–water mixture as the reaction solvent and K3PO4 as a base. This catalytic system might, however, cleave our acceptor groups from the molecular wires due to hydrolysis. We thus first tested the catalytic system for two ‘precursor’ systems (Br-BT-CHO and Br-CPDT-CHO) devoid of the respective acceptor groups. As shown in Table 1 (Entry 7) and Table 2 (Entry 4), the surface coverages increased tremendously. When the Pd(OAc)2/SPhos catalyst was used in combination with CsOAc base in a water-free THF–MeOH (5:1) mixture (Entry 8, Table 1 and Entry 6, Table 2), a comparable surface coverage was achieved. Hence, these conditions were also applied for the full molecular wire systems. As illustrated in Table 1 (Entry 8) and 2 (Entry 6), the overall coupling efficiency was noticeably higher when SPhos was used as the supporting ligand. A surface coverage of 0.58 and 0.56 ML was achieved for the Br-2T-Rho and Br-CPDT-Fur chromophores, respectively, the highest reported surface coverages so far for Suzuki cross-coupling on B:NCD films (Fig. 3). An increase in coverage of 17% is achieved despite the use of a less reactive Br–Ar function and a more complex molecular structure (0.56 ML for Br-CPDT-Fur) as compared to the results of Zhong and co-workers (0.41 ML for 2-(dicyanovinyl)-5-iodobithiophene).17 Moreover, the Pd catalyst usage is also quite low (3 mol%) as compared to standard Pd(PPh3)4, which requires a loading of 10 mol%.17
UPS was applied to probe the electronic properties of the valence band of the functionalized B:NCD samples. Fig. 4(a) and (c) show the UPS spectra of the Br-BT-Rho and Br-CPDT-Fur functionalized B:NCDs, respectively, compared to the respective pure molecular wire systems (drop-casted on an Au substrate). When the UPS intensity is plotted on a logarithmic scale, the weak valence band feature can be distinguished. The first valence band peak for both Br-BT-Rho and Br-BT-Rho functionalized B:NCD appears around 0.9 eV. On the other hand, for Br-CPDT-Fur and Br-CPDT-Fur functionalized B:NCD, the first valence band peak appears around 1.1 eV.
 | ||
| Fig. 4 (a and c) Comparison of the UPS spectra of the molecular wire-functionalized B:NCDs (blue line) and the pure molecular wires (drop-casted on an Au substrate) (black line) for (a) Br-BT-Rho and (c) Br-CPDT-Fur on an logarithmic scale. (b and d) Comparison of the UPS spectra of the molecular wire-functionalized B:NCDs (blue line) and a bare hydrogen-terminated B:NCD film (red line) for (b) Br-BT-Rho and (d) Br-CPDT-Fur on an logarithmic scale. Linear extrapolation of the valence band edge yields an estimate of the VBM the of B:NCD thin film. | ||
To determine the HOMO energy levels of both molecular wire systems, the UPS spectra of the functionalized diamond films were compared to bare hydrogen-terminated B:NCD thin films (Fig. 4b and d). By linear extrapolation of the hydrogen-terminated B:NCD thin film UPS data, the position of the valence band maximum (VBM) of diamond with respect to its Fermi level can be determined. It is approximately 0.6 eV below the (surface) Fermi level. Previous studies have shown that Ef-VBM is 0.25 eV in bulk B-doped diamond. However, it can increase up to 0.7 eV at the surface of a clean H-terminated diamond.26–29 Accordingly, the HOMO levels of the Br-BT-Rho and Br-CPDT-Fur functionalized diamond samples lie 0.3 and 0.5 eV below the VBM of B:NCD, respectively. These energy differences have implications on the possibility for hole transport between the B:NCD thin film and the functionalized molecular wires. Based on the principle of charge transfer in dye-sensitized solar cells with p-semiconducting photocathode (p-DSSCs), upon photogeneration of an exciton, the electron is transferred to an acceptor site and subsequently to the oxidized form of the electrolyte redox mediator. In turn, the hole is transferred from the donor site to the semiconductor electrode. Hence, a good match between the HOMO of the donor molecule and the valence band of the electrode is important for efficient hole injection. For B:NCD, the VBM is situated rather close to the HOMO of both molecular wires, which should favor hole injection from the light-harvesting chromophores to the B:NCD thin film.17,29
As a proof of concept, photoelectrochemical measurements were performed using either a hydrogen-terminated, Br-CPDT-Fur or Br-BT-Rho functionalized B:NCD film as the working electrode in a standard three-electrode electrochemical system (Fig. 5). After equilibration in darkness, the light source was switched on and off approximately every 10 s for several cycles. At time = 0, the electrode was in dark, and each light-on triggered the cathodic photocurrent. The process was fully reversible, and was stopped at time ≈ 300 s, when the electrode was relaxed again in dark. We observed an increased photocurrent intensity for both molecular wire functionalized B:NCD electrodes as compared to the control system. In accord with previous work,17 negative bias enhances the photocurrent considerably. This agrees well with the mechanism that, upon light generation of electron–hole pairs in the molecular wires, the separated electrons flow toward the MV2+ (dimethyl viologen) electron carrier and the holes toward the B:NCD electrode (at negative bias).16,17
 | ||
| Fig. 5 Photocurrent response of B:NCD (black line), Br-CPDT-Fur-B:NCD (blue lines), and Br-BT-Rho-B:NCD (red lines) electrodes in a 5 mM methyl viologen solution (in 0.1 M Na2SO4) at varying bias (vs. Ag/AgCl). White light illumination by a Xe-lamp (15 mW cm−2). Curves are offset for clarity, but the intensity scale is identical in all cases. | ||
Our photocurrents are comparable to those reported earlier for similar systems: Zhong et al.16 observed a photocurrent of ca. 150 nA cm−2 under white light (150 W halogen lamp) illumination of B:NCD sensitized by dicyanovinyl-bithiophene and C60-bithophene. Later on, the same group reported ca. 4–6 μA cm−2 under 1 sun illumination (AM1.5G; 100 mW cm−2).17 In general, photocurrents on sensitized diamond electrodes are very small compared to those observed for the state-of-art n-DSSCs with titania photoanode (≈20 mA cm−2 at 1 sun). Among other reasons, the difference can be rationalized in terms of surface morphology of the aforementioned semiconductor photoelectrodes. Early works on flat titania surface, sensitized with Ru–bipyridine complexes, reported photocurrents of several nA under white light (450 < λ/nm < 650) illumination from a 150 W Xe-lamp.30 In principle, the flat electrode cannot deliver external quantum efficiencies larger than ≈0.3%, whereas mesoporous electrodes can give values exceeding 90% for the same dye/electrolyte systems.31 The B:NCD films made by CVD are usually quite compact, with double-layer capacitances approaching those of flat surfaces (ca. 3 μF cm−2).32 Also our SEM image shown below (Fig. 6) confirms quite dense packing of the diamond crystals without any noticeable mesoporosity. Hence, surface engineering of the B:NCD photoanode is a straightforward task in the future development of diamond-based p-DSSCs.
 | ||
| Fig. 6 Typical SEM image of a B:NCD film. | ||
Conclusions
In summary, we have demonstrated that a proper choice of the catalytic system used for Suzuki cross-coupling can effectively increase the coupling efficiency, also on a diamond surface. Best results, leading to unprecedented surface loadings, were obtained for the Pd(OAc)2/SPhos combination. The resulting B:NCD films decorated with light-harvesting molecular dyes can potentially be applied for ‘all-carbon’ photovoltaic applications. Preliminary photoelectrochemical measurements using the novel molecular wire systems show photocurrent generation. More detailed studies concerning full devices and quantum efficiencies will be carried out and reported in near future.Experimental
Chemical reagents
Commercially available chemicals were purchased in their purest grade available and used without further purification. Sodium nitrite, sodium carbonate, sodium acetate, cesium acetate, cesium fluoride, potassium phosphate, palladium(II) acetate, 4-aminophenylboronic acid pinacol ester, rhodanine-3-acetic acid, tetrakis(triphenylphosphine)palladium(0), and tri(o-tolyl)phosphine were purchased from Sigma-Aldrich. 5-Bromo-5′-formyl-2,2′-bithiophene was purchased from TCI Europe N.V. 2-Dicyclohexylphosphino-2′,6′-dimethoxybiphenyl (SPhos) was purchased from Acros Organics. All solvents used for reactions and rinsing were of HPLC grade, unless otherwise stated. Water used for rinsing and electrochemical work was made with Type 1 Ultrapure water by Sartorius stedium Biotech.Synthesis and characterization
NMR chemical shifts (δ) were determined relative to the residual CHCl3 absorption (7.26 ppm) or the 13C resonance shift of CDCl3 (77.16 ppm). UV-Vis absorption spectra were recorded with an Agilent Cary 500 Scan UV-Vis-NIR spectrometer in a continuous run from 200 to 800 nm at a scan rate of 600 nm min−1. Electrochemical measurements were performed with an Eco Chemie Autolab PGSTAT 30 potentiostat/galvanostat using a three-electrode microcell equipped with a Pt wire working electrode, a Pt wire counter electrode and a Ag/AgNO3 reference electrode (Ag wire dipped in a solution of 0.01 M AgNO3 and 0.1 M NBu4PF6 in anhydrous MeCN). Samples were prepared in anhydrous CH2Cl2 containing 0.1 M NBu4PF6 and ferrocene was used as an internal standard. The respective products were dissolved in the electrolyte solution and degassed with argon prior to each measurement. To prevent air from entering the system, the experiments were carried out under argon. Cyclic voltammograms were recorded at a scan rate of 50 mV s−1. The HOMO–LUMO energy levels of Br-CPDT-Fur were determined using CV data. For the conversion of V to eV, the onset potentials of the first oxidation/reduction peaks were used and referenced to ferrocene/ferrocenium, which has an ionization potential of −4.98 eV vs. vacuum. This correction factor is based on a value of 0.31 eV for Fc/Fc+vs. SCE33a and a value of 4.68 eV for SCE vs. vacuum:33b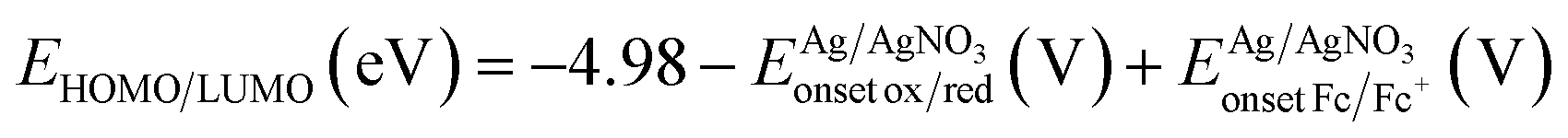 . To estimate the optical HOMO–LUMO gap, the wavelength at the intersection of the tangent line drawn at the low energy side of the (solution) absorption spectrum with the x-axis was used (ΔEopt (eV) = 1240/(wavelength in nm)).
. To estimate the optical HOMO–LUMO gap, the wavelength at the intersection of the tangent line drawn at the low energy side of the (solution) absorption spectrum with the x-axis was used (ΔEopt (eV) = 1240/(wavelength in nm)).
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH–), 7.04 (s 1H), 7.34 (s, 1H), 7.90 (d, 1H, J = 15.8 Hz, –CHCH–). 13C NMR (CDCl3): 9.3, 26.7, 30.3, 56.09, 56.12, 77.4, 95.6, 97.1, 110.5, 111.5, 111.7, 118.2, 125.3, 136.7, 140.6, 141.0, 147.6, 158.8, 161.4, 173.0, 176.1. UV-Vis (CHCl3): λmax = 585 nm, ε = 73100 L mol−1 cm−1, ΔEopt = 1.85 eV.
000 ppm B/C to CH4 to ensure good electrical conductivity.1b Based on previous experiments,35 this ratio corresponds to a boron film concentration 1021 cm−3. Sheet resistance (Rs), as measured by a four-point probe measurement, was 207 Ω sq−1, which agrees well with the typical value for B:NCD grown under these conditions.3 Prior to the diamond growth, the fused silica substrates were cleaned for 15 min each in RCA 1 (30% NH3 + 30% H2O2 + H2O; 1:1:5) and RCA 2 (37% HCl + 30% H2O2 + H2O; 1:1:5) solutions at 90 °C. Following this, the cleaned fused silica substrates were seeded with nanodiamond powder in water to improve the nucleation density. After deposition, the diamond samples were allowed to cool down in the reactor for 30 min under vacuum. To remove any graphitic layers, the as-deposited diamond films were boiled in an acidic mixture of 99% H2SO4 + 30% HNO3 (3:1) at 90 °C for 30 min. After rinsing and sonicating with plenty of deionized water, the diamond samples were subjected to hydrogenation. Fig. 6 displays a scanning electron microscopy (SEM) image of a typical as-grown B:NCD thin film.
CH–), 7.04 (s 1H), 7.34 (s, 1H), 7.90 (d, 1H, J = 15.8 Hz, –CHCH–). 13C NMR (CDCl3): 9.3, 26.7, 30.3, 56.09, 56.12, 77.4, 95.6, 97.1, 110.5, 111.5, 111.7, 118.2, 125.3, 136.7, 140.6, 141.0, 147.6, 158.8, 161.4, 173.0, 176.1. UV-Vis (CHCl3): λmax = 585 nm, ε = 73100 L mol−1 cm−1, ΔEopt = 1.85 eV.
000 ppm B/C to CH4 to ensure good electrical conductivity.1b Based on previous experiments,35 this ratio corresponds to a boron film concentration 1021 cm−3. Sheet resistance (Rs), as measured by a four-point probe measurement, was 207 Ω sq−1, which agrees well with the typical value for B:NCD grown under these conditions.3 Prior to the diamond growth, the fused silica substrates were cleaned for 15 min each in RCA 1 (30% NH3 + 30% H2O2 + H2O; 1:1:5) and RCA 2 (37% HCl + 30% H2O2 + H2O; 1:1:5) solutions at 90 °C. Following this, the cleaned fused silica substrates were seeded with nanodiamond powder in water to improve the nucleation density. After deposition, the diamond samples were allowed to cool down in the reactor for 30 min under vacuum. To remove any graphitic layers, the as-deposited diamond films were boiled in an acidic mixture of 99% H2SO4 + 30% HNO3 (3:1) at 90 °C for 30 min. After rinsing and sonicating with plenty of deionized water, the diamond samples were subjected to hydrogenation. Fig. 6 displays a scanning electron microscopy (SEM) image of a typical as-grown B:NCD thin film.
The surface coverage with the thiophene moieties was estimated using the same method as used in ref. 17. This is done by calibrating the XPS S 2p signal of the thiophene moieties against the measured XPS S 2p intensity of a dodecanethiol SAM on Au, where the coverage of the latter was assumed to be 100% (1 ML) (Fig. 7). Several samples of dodecanethiol SAMs were prepared on Au, i.e. at different functionalization times, to make sure the same S 2p signal was obtained by XPS. The S intensity in dodecanethiol has been corrected for the attenuation factor due to the carbon chain.

 | ||
| Fig. 7 XPS spectrum showing the S 2p signal of dodecanethiol on a Au surface. | ||
Acknowledgements
This work was financially supported by the Special Research Fund of Hasselt University, the Research Foundation Flanders (FWO) (G.0555.10N), and the EU FP7 Collaborative Project “MOLESOL” (no. 256617). X.L. acknowledges support from The Swedish Research Council Linnaeus grant LiLi-NFM. L.K. acknowledges support from the Grant Agency of the Czech Republic (contract no. 13-37383S). We thank Huguette Penxten for the cyclic voltammetry measurements.Notes and references
- (a) O. Williams and M. Nesladek, in Physics and Applications of CVD Diamond, ed. S. Koizumi, C. Nebel and M. Nesladek, Wiley-VCH, Weinheim, Germany, 2008 Search PubMed; (b) S. D. Janssens, P. Pobedinskas, J. Vacik, V. Petrakova, B. Ruttens, J. D'Haen, M. Nesladek, K. Haenen and P. Wagner, New J. Phys., 2011, 13, 083008 CrossRef.
- O. A. Williams, M. Nesladek, M. Daenen, S. Michaelson, A. Hoffman, E. Osawa, K. Haenen and R. B. Jackman, Diamond Relat. Mater., 2008, 17, 1080 CrossRef CAS PubMed.
- C. H. Y. X. Lim, Y. L. Zhong, S. Janssens, M. Nesladek and K. P. Loh, Adv. Funct. Mater., 2010, 20, 1313 CrossRef CAS.
- (a) R. S. Balmer, J. R. Brandon, S. L. Clewes, H. K. Dhillon, J. M. Dodson, I. Friel, P. N. Inglis, T. D. Madgwick, M. L. Markham, T. P. Mollart, N. Perkins, G. A. Scarsbrook, D. J. Twitchen, A. J. Whitehead, J. J. Wilman and S. M. Woollard, J. Phys.: Condens. Matter, 2009, 21, 36422 CrossRef PubMed; (b) A. Kirste, G. Schnakenburg, F. Stecker, A. Fischer and S. R. Waldvogel, Angew. Chem., Int. Ed., 2010, 49, 971 CrossRef CAS PubMed; (c) Q. Wang, P. Subramanian, M. S. Li, W. S. Yeap, K. Haenen, Y. Coffinier, R. Boukherroub and S. Szunerits, Electrochem. Commun., 2013, 34, 286 CrossRef CAS PubMed; (d) S. Inagi, H. Nagai, I. Tomita and T. Fuchigami, Angew. Chem., Int. Ed., 2013, 52, 6616 CrossRef CAS PubMed; (e) T. L. Read, E. Bitziou, M. B. Joseph and J. V. Macpherson, Anal. Chem., 2014, 86, 367 CrossRef CAS PubMed; (f) K. Nakata, T. Ozaki, C. Terashima, A. Fujishima and Y. Einaga, Angew. Chem., Int. Ed., 2014, 53, 871 CrossRef CAS PubMed.
- X. Y. Wang, E. C. Landis, R. Franking and R. J. Hamers, Acc. Chem. Res., 2010, 43, 1205 CrossRef CAS PubMed.
- X. Wang, S. Kurihara, M. Hasegawa, A. R. Ruslinda and H. Kawarada, Jpn. J. Appl. Phys., 2012, 51, 090125 CrossRef.
- D. Belanger and J. Pinson, Chem. Soc. Rev., 2011, 40, 3995 RSC.
- J. Lahann, in Click Chemistry for Biotechnology and Materials Science, ed. J. Lahann, John Wiley & Sons Ltd., West Sussex, 2009, pp. 1–8 Search PubMed.
- N. Miyaura and A. Suzuki, Chem. Rev., 1995, 95, 2457 CrossRef CAS.
- P. Bujak, I. Kulszewicz-Bajer, M. Zagorska, V. Maurel, I. Wielgus and A. Pron, Chem. Soc. Rev., 2013, 42, 8895 RSC.
- A. Pron, P. Gawrys, M. Zagorska, D. Djurado and R. Demadrille, Chem. Soc. Rev., 2010, 39, 2577 RSC.
- (a) J. C. Bijleveld, M. Shahid, J. Gilot, M. M. Wienk and R. A. J. Janssen, Adv. Funct. Mater., 2009, 19, 3262 CrossRef CAS; (b) D. C. Watters, H. Yi, A. J. Pearson, J. Kingsley, A. Iraqi and D. Lidzey, Macromol. Rapid Commun., 2013, 34, 1157 CrossRef CAS PubMed.
- W. Wu, L. Huang, L. Xiao, Q. Huang, R. Tang, C. Ye, J. Qin and Z. Li, RSC Adv., 2012, 2, 6520 RSC.
- (a) F. Cheng and A. Adronov, Chem. Mater., 2006, 18, 5389 CrossRef CAS; (b) J. Gao, F. Bao, Q. Zhu, Z. Tan, T. Chen, H. Cai, C. Zhao, Q. Cheng, Y. Yang and R. Ma, Polym. Chem., 2013, 4, 1672 RSC.
- T. Hasobe, H. Imahori, H. Yamada, T. Sato, K. Ohkubo and S. Fukuzumi, Nano Lett., 2003, 3, 409 CrossRef CAS.
- Y. L. Zhong, K. P. Loh, A. Midya and Z. K. Chen, Chem. Mater., 2008, 20, 3137 CrossRef CAS.
- Y. L. Zhong, A. Midya, Z. Ng, Z. K. Chen, M. Daenen, M. Nesladek and K. P. Loh, J. Am. Chem. Soc., 2008, 130, 17218 CrossRef CAS PubMed.
- W. S. Yeap, S. Chen and K. P. Loh, Langmuir, 2009, 25, 185 CrossRef CAS PubMed.
- (a) J. Peet, J. Y. Kim, N. E. Coates, W. L. Ma, D. Moses, A. J. Heeger and G. C. Bazan, Nat. Mater., 2007, 6, 497 CrossRef CAS PubMed; (b) R. Li, J. Liu, N. Cai, M. Zhang and P. Wang, J. Phys. Chem. B, 2010, 114, 4461 CrossRef CAS PubMed; (c) H. N. Tsao, D. M. Cho, I. Park, M. R. Hansen, A. Mavrinskiy, D. Y. Yoon, R. Graf, W. Pisula, H. W. Spiess and K. Müllen, J. Am. Chem. Soc., 2011, 133, 2605 CrossRef CAS PubMed; (d) S. Van Mierloo, A. Hadipour, M.-J. Spijkman, N. Van den Brande, B. Ruttens, J. Kesters, J. D'Haen, G. Van Assche, D. M. de Leeuw, T. Aernouts, J. Manca, L. Lutsen, D. Vanderzande and W. Maes, Chem. Mater., 2012, 24, 587 CrossRef CAS; (e) P. Gao, H. N. Tsao, M. Graetzel and M. K. Nazeeruddin, Org. Lett., 2012, 14, 4330 CrossRef CAS PubMed; (f) M. Marszalek, S. Nagane, A. Ichake, R. Humphry-Baker, V. Paul, S. M. Zakeeruddin and M. Grätzel, RSC Adv., 2013, 3, 7921 RSC; (g) J. H. Delcamp, Y. Shi, J.-H. Yum, T. Sajoto, E. Dell'Orto, S. Barlow, M. K. Nazeeruddin, S. R. Marder and M. Graetzel, Chem.–Eur. J., 2013, 19, 1819 CrossRef CAS PubMed.
- (a) W. Vanormelingen, P. Verstappen, V. Maes, D. Bevk, L. Lutsen, D. Vanderzande and W. Maes, Synlett, 2013, 24, 2389 CrossRef CAS PubMed; (b) S. Van Mierloo, P. J. Adriaensens, W. Maes, L. Lutsen, T. J. Cleij, E. Botek, B. Champagne and D. J. Vanderzande, J. Org. Chem., 2010, 75, 7202 CrossRef CAS PubMed.
- A. Suzuki, Chem. Commun., 2005, 4759 RSC.
- (a) K. L. Billingsley, T. E. Barder and S. L. Buchwald, Angew. Chem., Int. Ed., 2007, 46, 5359 CrossRef CAS PubMed; (b) F.-S. Han, Chem. Soc. Rev., 2013, 42, 5270 RSC.
- T. E. Barder, S. D. Walker, J. R. Martinelli and S. L. Buchwald, J. Am. Chem. Soc., 2005, 127, 4685 CrossRef CAS PubMed.
- J. F. Hartwig, Acc. Chem. Res., 2008, 41, 1534 CrossRef CAS PubMed.
- D. F. Zhou, N. Cai, H. J. Long, M. Zhang, Y. H. Wang and P. Wang, J. Phys. Chem. C, 2011, 115, 3163 CAS.
- C. Bandis and B. B. Pate, Phys. Rev. B: Condens. Matter Mater. Phys., 1995, 52, 12056 CrossRef CAS.
- L. Diederich, O. M. Küttel, P. Aebi and L. Schlapbach, Surf. Sci., 1998, 418, 219 CrossRef CAS.
- F. J. Himpsel, J. F. van der Veen and D. E. Eastman, Phys. Rev. B: Condens. Matter Mater. Phys., 1980, 22, 1967 CrossRef CAS.
- I. Zegkinoglou, P. L. Cook, P. S. Johnson, W. L. Yang, J. H. Guo, D. Pickup, R. Gonzalez-Moreno, C. Rogero, R. E. Ruther, M. L. Rigsby, J. E. Ortega, R. J. Hamers and F. J. Himpsel, J. Phys. Chem. C, 2012, 116, 13877 CAS.
- M. P. Dare-Edwards, J. B. Goodenough, A. Hamnett, K. R. Seddon and R. D. Wright, Discuss. Faraday Soc., 1980, 70, 285 RSC.
- L. Kavan, Chem. Rec., 2012, 12, 131 CrossRef CAS PubMed.
- A. Fujishima, Y. Einaga, T. N. Rao and D. A. Tryk, Diamond Electrochemistry, ed. A. Fujishima, Y. Einaga, T. N. Rao and D. A. Tryk, Elsevier, Amsterdam, 2005, pp. 556–574 Search PubMed.
- (a) A. J. Bard and L. R. Faulkner, Electrochemical Methods: Fundamentals and Applications, John Wiley & Sons Inc., New York, 2001 Search PubMed; (b) S. Trasatti, Pure Appl. Chem., 1986, 58, 955 CAS.
- S. Ermer, S. M. Lovejoy, P. V. Bedworth, D. S. Leung, H. B. Warren, J. A. Epstein, D. G. Girton, L. S. Dries, R. E. Taylor, R. R. Barto, W. Eades, T. E. Van Eck, A. S. Moss and W. W. Anderson, Adv. Funct. Mater., 2002, 12, 605 CrossRef CAS.
- W. Gajewski, P. Achatz, O. A. Williams, K. Haenen, E. Bustarret, M. Stutzmann and J. A. Garrido, Phys. Rev. B: Condens. Matter Mater. Phys., 2009, 79, 045206 CrossRef.
- P. E. Laibinis, C. D. Bain and G. M. Whitesides, J. Phys. Chem., 1991, 95, 7017 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: 1H and 13C NMR and UV-Vis spectra of Br-CPDT-Fur. See DOI: 10.1039/c4ra04740k |
| ‡ W.S.Y., L.L., W.M., and K.H. conceived the concept and designed the experiments. W.S.Y. performed the diamond growth and surface functionalization in K.H., W.M., and D.V.'s labs. Surface studies were carried out in M.F.'s lab. D.B. performed the synthesis of the Br-CPDT-Fur and Br-BT-Rho dyes. L.X. performed the photoelectron spectroscopy measurements. A.P. and H.K. performed the photoelectrochemical measurements. W.S.Y., D.B., L.X., W.M., L.K., and K.H. discussed the data and wrote the manuscript. All authors provided comments on the manuscript. |
| This journal is © The Royal Society of Chemistry 2014 |