
Subtle backbone modifications control the interpenetration of dibenzosuberone-based coordination cages†
Abstract
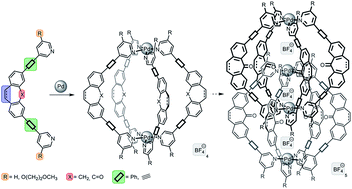
In order to gain a better understanding of the formation of interpenetrated double-cages [Pd4L8] from banana-shaped ligands and square-planar metal cations, our previously reported dibenzosuberone-based ligand was synthetically modified. We show that the formation of double-cages tolerates a wide range of suberone backbone modifications, as long as the ketone functionality is preserved. Reduction of this group to a CH2-bridge allowed us to form a monomeric [Pd2L4] cage, instead. PM6 and DFT calculations were performed to study the role of the carbonyl group in the formation of double-cages yielding results that are in good agreement with the experiments.
- This article is part of the themed collection: Supramolecular chemistry: self-assembly and molecular recognition
 Please wait while we load your content...
Please wait while we load your content...

