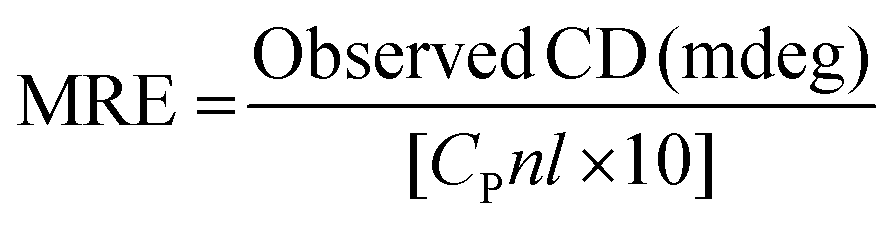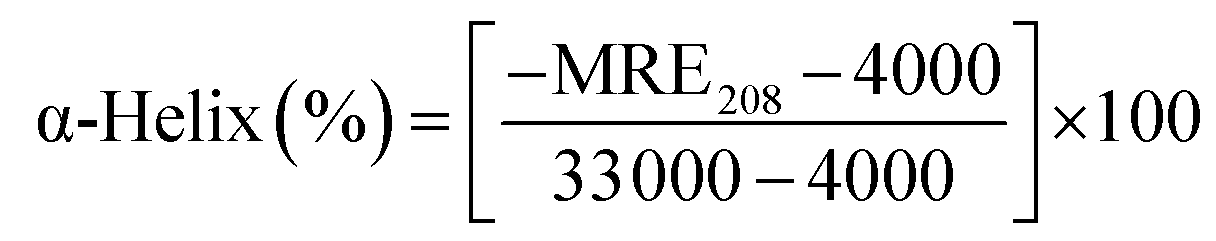DOI:
10.1039/C4RA04630G
(Paper)
RSC Adv., 2014,
4, 36267-36281
In vitro investigation of domain specific interactions of phenothiazine dye with serum proteins by spectroscopic and molecular docking approaches
Received
16th May 2014
, Accepted 6th August 2014
First published on 6th August 2014
Abstract
In the present study the interaction of the chemotherapeutic agent, Azure A (AZA) with Human Serum Albumin (HSA) and Bovine Serum Albumin (BSA) was investigated by multi spectroscopic and molecular docking methods. The influence of inner filter effect (IFE) on the emission quenching of HSA/BSA at low concentration of AZA (absorption value <0.1) suggested the need to employ an IFE correction factor even for the low concentration regime. The emission titration experiments of HSA/BSA with AZA revealed the formation of AZA–HSA/BSA complexes. The binding parameters calculated from corrected emission intensities showed that AZA binds to HSA/BSA with moderately strong binding affinities. The negative free energy obtained for the binding of AZA with HSA/BSA indicated that the complexation process is spontaneous. The results from site maker competitive experiments with specific site markers revealed that the probable binding location of AZA is located near site I of HSA/BSA. An AutoDock based molecular docking approach was utilized to characterize the binding models of AZA–HSA/BSA complexes. The free energy calculations for the most stable conformer from molecular docking studies were utilized to examine the energy contributions and the role of various amino acid residues of HSA/BSA in AZA binding. The results of site-competitive replacement experiments with specific site markers and molecular docking simulation studies unambiguously helped us to conclude that AZA binds to site I of HSA/BSA. Constant wavelength synchronous emission, excitation–emission matrix (three-dimensional) emission, absorption and circular dichroism spectroscopic techniques have been exploited to unravel AZA induced tertiary and secondary conformational changes of HSA/BSA.
Introduction
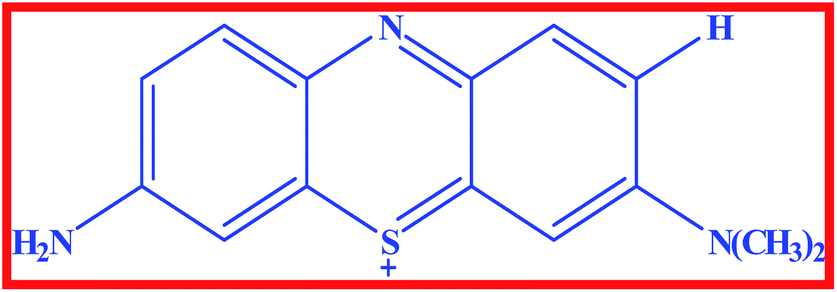
In recent years, several organic dye molecules are being increasingly used for clinical and medicinal purposes. Among them, phenothiazinium dyes have widespread applications in the fields of medicine, solar cells, biosensors, etc.1 These dyes are cationic compounds with high redox potential that interact with visible light inactivating several kinds of pathogenic agents in fresh plasma.2 This group of dyes present great reactivity with proteins and lipoproteins (cell membranes) and nucleic acids. These cationic compounds have limited capability to permeate the cell membrane as a function of their elevated hydrophilic character.1 Moreover, it present significant action against encapsulated viruses and some viruses without capsule, such as parvovirus B19.1 Azure A (AZA) is an asymmetric dimethyl thionine dye belonging to phenothiazinium group (Scheme 1) and it has significant biological importance such as anti-malarial effects, diagnosis of amyloid accumulation related diseases, and reducing the extent of cardiac arrhythmias.3,4 AZA has been shown to possesses good photo chemotherapeutic activity against carcinomas and in the treatment of angiogenic diseases.5 Besides, it has important applications in electro catalysis, electro chromic devices, solar energy cells, optical sensors and photovoltaic cells.6,7 Besides AZA shows good affinity towards DNA poly nucleotides and it undergoes both electrostatic and intercalative mode of binding with DNA.8
 |
| | Scheme 1 Structure of Azure A. | |
Serum albumins are extensively used in biophysical and biochemical studies as a model system for protein folding, aggregation and drug delivery.9 Serum albumins are one of the most abundant proteins present in the blood plasma, which assists in the disposition and transportation of various exogenous and endogenous ligands to specific targets.9 Albumins are the principal bio-macromolecules responsible for the maintenance of colloid-blood pressure needed for proper distribution of body fluids between intravascular compartments and body tissues.10,11 They also act as plasma carrier through nonspecific binding with several hydrophobic steroid hormones across organ–circulatory interfaces such as the liver, intestine, kidney and brain.12,13
Structural aspects of human serum albumin (HSA) and bovine serum albumin (BSA) have been reasonably well explored. The primary structure of these proteins have about 580 amino acid residues which assume the solid equilateral triangular shape with sides ∼80 Å and depth ∼30 Å. HSA/BSA proteins are characterized by low tryptophan (Trp) content and a high content of cystine stabilizing a series of nine loops. The secondary structure of these serum albumins is constituted of ∼67% helix content with six turns and 17 disulfide bridges.14,15 Human and bovine serum albumins display approximately 80% sequence homology and a repeating pattern of disulfides that is strictly conserved. The primary structure of HSA consists of 585 amino acids and its amino acid sequence contains 18 tyrosines (Tyr), 6 methionines, 1 tryptophan (Trp-214), 17 disulfide bridges and one free thiol group. The disulfides are positioned in a repeating series of nine loop like structures centered on eight sequential Cys–Cys pairs. In the case of BSA, the primary structure is composed of 583 amino acid residues consisting of two Trp residues (Trp-134 and Trp-213). From the determined crystallographic structure of HSA, it is proposed that the single Trp residue (Trp-214) is located in IIA binding site. In the case of BSA, Trp-213 is located in a similar hydrophobic microenvironment as the single Trp-214 in HSA (subdomain IIA), whereas, Trp-134 is more exposed to solvent and it is localized in the subdomain IA.16,17
The tertiary structure of HSA/BSA is composed of three domains I, II and III. Each domain is constituted by two sub domains namely A and B. Since domains II and III share a common interface, binding of a probe to domain III leads to conformational changes affecting the binding affinities to domain II. The principal ligand binding sites are located in sub domain IIA, IIIA and it appears that in the HSA/BSA these sites are homologous, although, they differ in affinities. The specific delivery of ligands by serum albumins originates from the presence of two major and structurally selective binding sites, namely, Sudlow's site I and site II, which are located in three homogeneous domains that form a heart-shaped structure. The binding affinity offered by site I is mainly through hydrophobic interaction, while site II involves a combination of hydrophobic, hydrogen bonding and electrostatic interactions.14–19 It has been reported that molecules possessing higher affinity for serum albumins and showing preferential binding at site II are shown to exhibit efficient photodynamic therapeutic applications.20 Therefore, in-depth analysis on the site selective binding interactions of biologically active ligands with serum albumins is essential for designing efficient drugs for medical purposes.21 It is widely accepted in the pharmaceutical industry that the overall distribution, metabolism and efficacy of many drugs can be altered based on their affinity to serum albumin. In addition, many promising new drugs are rendered ineffective because of their unusual high affinity for this abundant protein.21 Obviously, the understanding of chemistry involved in the various classes of pharmaceutical interactions with serum albumins can suggest new approaches to drug therapy and design. Consequently, the present investigation is designed to focus on deciphering the binding interaction of the biologically active AZA with HSA/BSA by means of steady-state emission spectroscopy. To unravel the effect of AZA binding on the secondary structure of serum proteins constant wavelength synchronous emission, three dimensional emission, absorption and circular dichroism studies were carried out. We have also employed computational approaches to determine the exact binding location of AZA within HSA/BSA.
Materials and methods
Materials
Human Serum Albumin (HSA, fraction V) was purchased from Sigma-Aldrich, USA and Bovine Serum Albumin (BSA, fraction V), purchased from Himedia, India were used without further purification. Ibuprofen and phenylbutazone were obtained from TCI chemicals, Japan and were used as received. Azure A (AZA) was obtained from SD-Fine Chemicals, India. It was purified by column chromatography on neutral alumina using ethanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :benzene (7:3 v/v) containing 0.4% glacial acetic acid and then recrystallized from ethanol.22 Stock solutions of HSA/BSA were prepared by using phosphate buffer solution (PBS) of pH = 7.2. The concentration of HSA/BSA was measured spectrophotometrically by reported procedure.23 All other reagents were of analytical grade and were used as received. Water used in this experiment was doubly distilled over alkaline potassium permanganate using an all glass apparatus.
:benzene (7:3 v/v) containing 0.4% glacial acetic acid and then recrystallized from ethanol.22 Stock solutions of HSA/BSA were prepared by using phosphate buffer solution (PBS) of pH = 7.2. The concentration of HSA/BSA was measured spectrophotometrically by reported procedure.23 All other reagents were of analytical grade and were used as received. Water used in this experiment was doubly distilled over alkaline potassium permanganate using an all glass apparatus.
Absorption and emission spectral measurements
Absorption spectral measurements were carried out using JASCO V-630 UV-Visible spectrophotometer. Quartz cuvettes of path length 1 cm were used to record the absorption spectra. The emission spectral studies were performed with JASCO FP-6600 spectrofluorometer. All the titration experiments were carried out by adding appropriate amount of AZA to 1 mL of HSA/BSA solution in a 5 mL standard measuring flask in sequence and then made up to the mark with PBS solution. The solution was mixed uniformly and allowed to equilibrate for 15 minutes and the homogeneous solution was taken in a quartz cuvette (1 cm) before recording the spectra. HSA/BSA was excited at 295 nm and the emission was monitored in the region between 300 nm to 500 nm. The emission and excitation slit widths used throughout the experiment were 5 nm and 2 nm, respectively. The synchronous emission spectra were recorded at Δλ = 15 nm and Δλ = 60 nm. Three-dimensional emission spectra were recorded over the range of 200 nm to 500 nm at a scanning rate of 2000 nm min−1. All the measurements were carried out at room temperature (25 °C). Stock solutions of serum albumins and AZA were always freshly prepared before use.
Circular dichroism measurements
Circular dichroism (CD) measurements were performed with a JASCO-180 spectropolarimeter using a 0.1 cm path length quartz cell. The CD spectra were recorded in the range of 200–300 nm with 0.1 nm step resolution and averaged over two scans at a speed of 50 nm min−1. All observed spectra were baseline subtracted for buffer solution and the α-helical content was calculated on the basis of change of molar ellipticity value.
Molecular docking studies
Molecular docking experiments were performed using the docking software AutoDock 4.2 along with the AutoDock Tools (ADT). The crystallographic coordinates of AZA was obtained from the PubChem database. The native structures of HSA (PDB id: 1AO6) and BSA (PDB id: 4F5S) were retrieved from the Protein Data Bank. As required for Lamarckian Genetic Algorithm docking compilation, all water molecules were removed from the native structure of BSA/HSA with subsequent addition of hydrogen atoms followed by the calculation of Gasteiger charges. The grid sizes along the x-, y-, z-axes was set to 60 Å, 60 Å and 60 Å for HSA and 44 Å, 44 Å, 44 Å for BSA, respectively. The grid spacing was set as 0.403 Å for both proteins. During the docking process the whole sub domain IIA of HSA/BSA was encompassed by setting the grid center along the x-, y-, z-axes to 34.016 Å, 42.121 Å, 50.644 Å, for HSA and BSA, the axes values was set to −4.011 Å, 24.628 Å and 106.569 Å. The AutoDocking parameters used were, Genetic Algorithm (GA) population = 150; maximum number of energy evaluations = 250000 and GA crossover mode of two points. For each docking simulation, 25 different conformers were generated. The docked conformations were visualized using PyMOL software package. The conformation with lowest binding free energy was used for further analysis.24
Results and discussion
Emission spectral studies of HSA/BSA with AZA
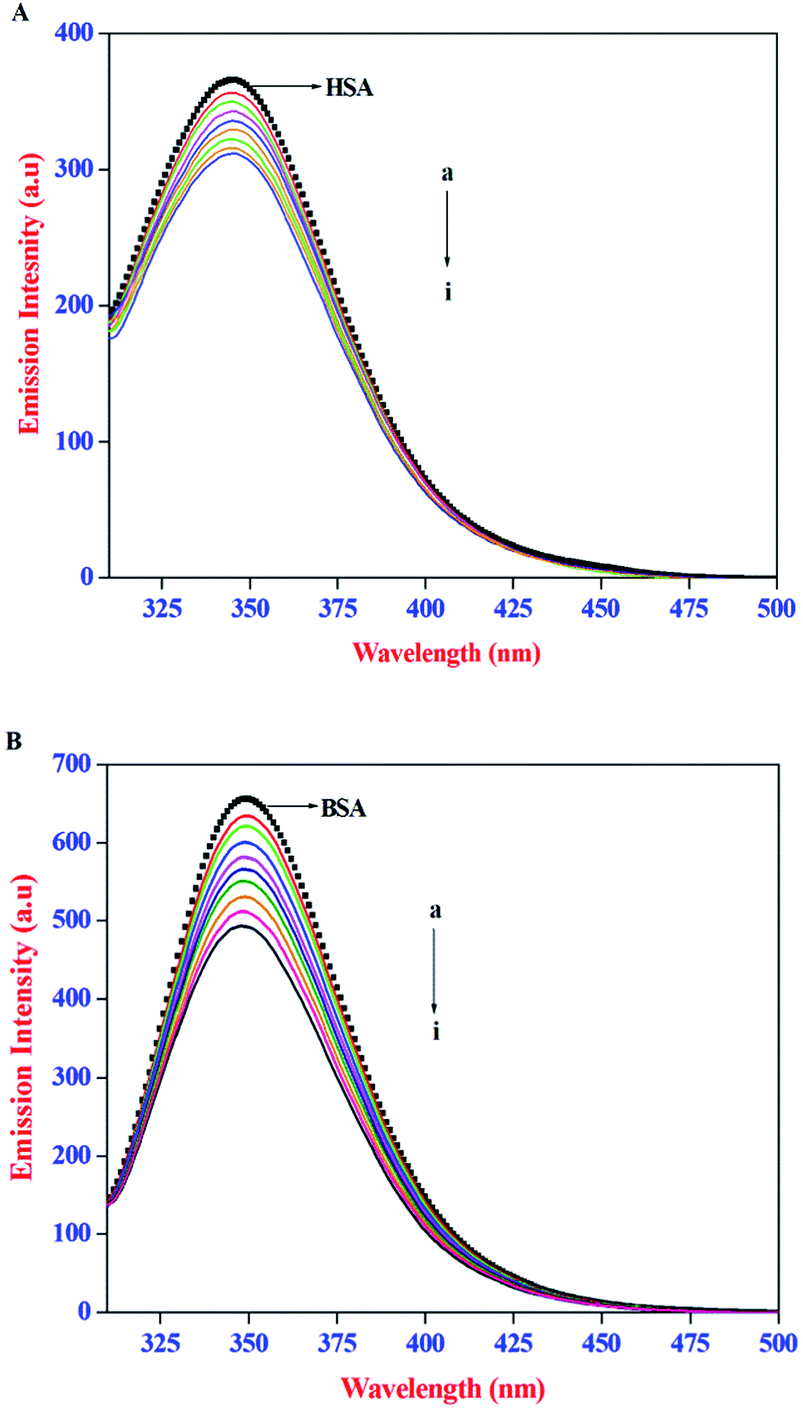
The intrinsic emission property of proteins, arise mainly from the tryptophan (Trp), tyrosine (Tyr), and phenylalanine (Phe) residues. It is well established that the protein–ligand interactions often lead to change in the intrinsic emission property of proteins. As a result, emission spectral techniques are widely employed to study the binding interactions of small molecules to protein. In the present study, the effect of AZA on the intrinsic emission property of HSA/BSA was monitored using emission spectroscopy. The emission spectra recorded for HSA/BSA with different concentrations of AZA are shown in Fig. 1(A) and (B). The emission spectrum of HSA/BSA in the absence of AZA shows an emission maximum at 348 nm, when excited at 295 nm. The excitation wavelength of 295 nm was chosen to avoid any contribution from Tyr residue and the resulting emission spectrum is exclusively ascribed to the intrinsic Trp fluorophore.25 It was observed from Fig. 1(A) and (B), that with the addition of increasing concentrations of AZA to HSA/BSA solution, the emission intensity of HSA/BSA showed a progressive decrease. Since the emission spectrum of HSA/BSA depends on the degree of exposure of the Trp residues to the solvent polarity and upon its proximity to specific quenching groups, the observed decrease in emission intensity could be attributed to the binding of AZA with HSA/BSA.26 The above interpretations showed that the binding site of AZA on HSA/BSA is most probably located near the Trp residues of HSA (Trp-214) and BSA (Trp-213).27 It is well known that quenching of fluorescent macromolecule can occur due to IFE (Inner Filter Effect), collisional quenching and binding-related quenching. The binding-related quenching is further subdivided into ground-state complex formation between the ligand and the macromolecule, excited-state quenching in the complex (e.g., energy transfer), or structural changes around the fluorophores.28 Therefore, in the present case the exact mechanism of emission quenching in HSA/BSA by AZA was investigated.
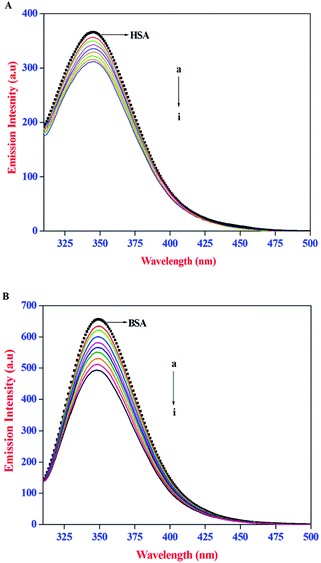 |
| | Fig. 1 Emission spectra of (A) HSA (1.00 × 10−6 mol dm−3) and (B) BSA (1.00 × 10−6 mol dm−3) at various AZA concentrations. [AZA]: (a) 0, (b) 2.00 × 10−7, (c) 4.00 × 10−7, (d) 6.00 × 10−7, (e) 8.00 × 10−7, (f) 10.00 × 10−7, (g) 12.00 × 10−7, (h) 14.00 × 10−7 and (i) 16.00 × 10−7 mol dm−3. | |
Influence of IFE on emission quenching of HSA/BSA by AZA
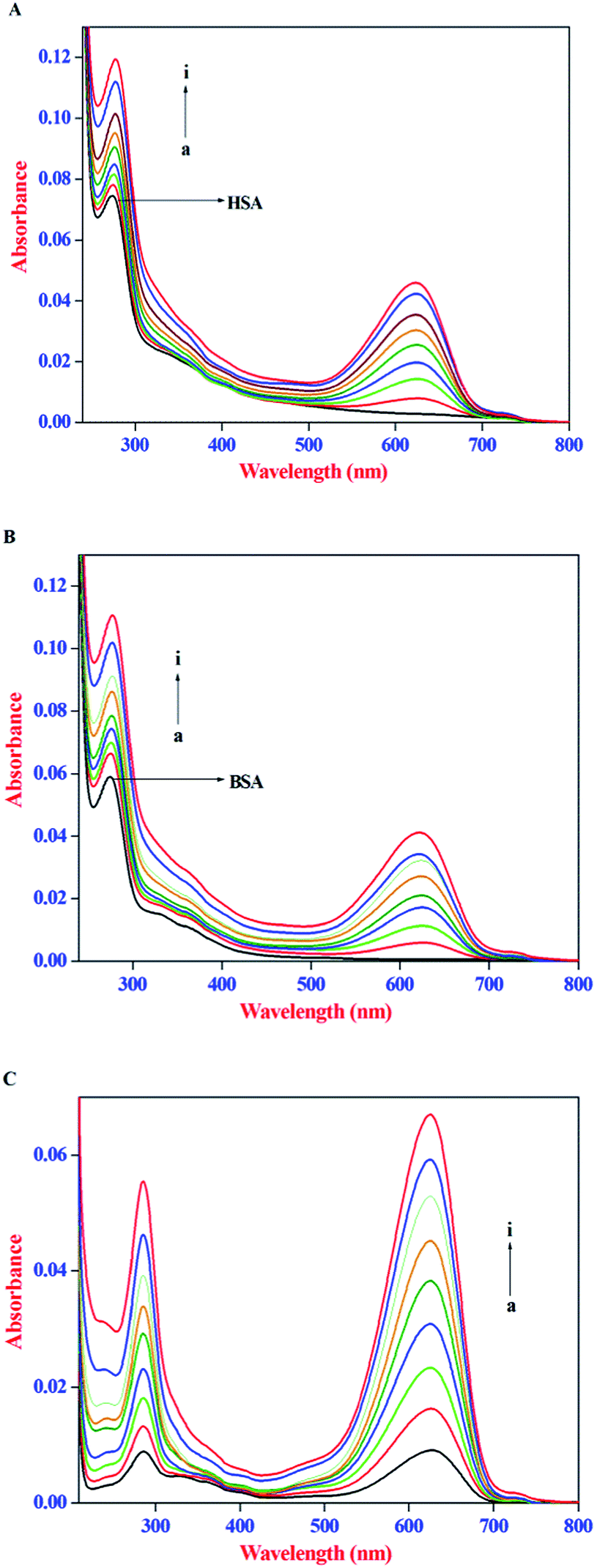
IFE refers to the absorption (or optical dispersion) of light at the excitation or emission wavelength by the compounds present in the solution. When the ligand molecules used in the protein binding studies possess strong absorption at the excitation wavelength of the protein, less light reaches the centre of the sample and thus the emission intensity of the protein will be reduced, whereas, a strong absorption at the emission wavelength of protein would reduce the emitted light that reaches the detector. Therefore, in the present investigation, the IFE of AZA on the emission intensity of both HSA and BSA was evaluated. The absorption spectra of HSA/BSA in the presence of increasing concentrations of AZA and absorption spectra of AZA alone in the concentration range of 0 to 16.00 × 10−7 mol dm−3 are shown in Fig. 2(A–C). As shown in Fig. 2(A) and (B), in the absence of AZA, both HSA and BSA exhibit an absorption maxima at 278 nm which mainly originates from aromatic residues and disulfide bonds in the protein.26 It is clear from the absorption spectra that with the addition of increasing concentrations of AZA, the absorption intensity of HSA/BSA at 278 nm increased and the absorption maximum red shifted from 278 nm to 283 nm. In several literature reports, the role of ligand absorption values of less than 0.1 is ignored and in turn the influence of IFE on the emission intensity of protein is considered to be negligible.29,30 However, it has been reported that a change in absorption equal to 0.03 corresponds to a 3% reduction in emission intensity of protein.31 Therefore, it is necessary to correct the observed emission intensity for IFE even for absorption values <0.1. It can be seen from Fig. 2(C) that within the investigated concentration range, the absorption intensity value of AZA at the excitation (295 nm) and emission wavelength (348 nm) of HSA/BSA is in the range of 0.001 to 0.05. This clearly indicates the possible occurrence of the IFE. The considerable absorption value of AZA at 295 nm will reduce the amount of excitation radiation that reaches the Trp residues. Simultaneously, it can also absorb some radiation emitted by the Trp at 348 nm, leading to a decrease in the emission intensity of HSA/BSA. As a result, the influence of IFE on emission quenching of HSA/BSA has to be removed before conducting an analysis of the quenching mechanism. Similar kind of observation has been reported for the binding interactions of β-conglycinin and glycinin with vitamin B12.32
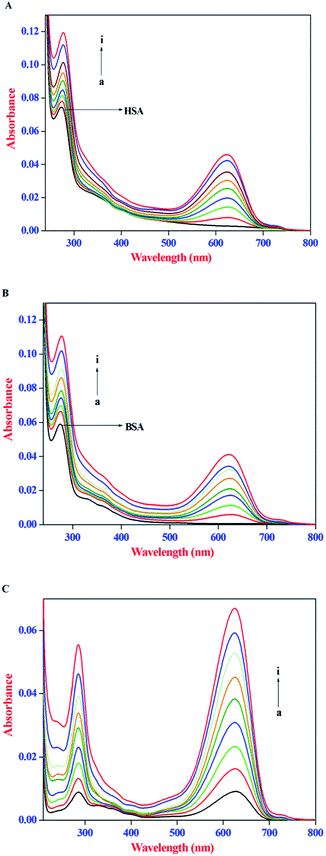 |
| | Fig. 2 Absorption spectra of (A) HSA (1.00 × 10−6 mol dm−3), (B) BSA (1.0 × 10−6 mol dm−3) in the absence and presence of AZA. (C) AZA alone. Conditions: [AZA]: (a) 0, (b) 2.00 × 10−7, (c) 4.00 × 10−7, (d) 6.00 × 10−7, (e) 8.00 × 10−7, (f) 10.00 × 10−7, (g) 12.00 × 10−7, (h) 14.00 × 10−7 and (i) 16.00 × 10−7 mol dm−3. | |
The correction of emission intensity can be achieved by measuring the absorbance values at the excitation and emission wavelength for each concentration of AZA and then multiplying the observed emission intensity value. Accordingly, the emission intensities were corrected using the relationship, eqn (1):28
| |
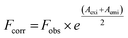 | (1) |
where,
Fcorr and
Fobs are the corrected and observed emission intensities, respectively.
Aexi and
Aemi are the solution absorbance at the excitation and emission wavelengths, respectively.
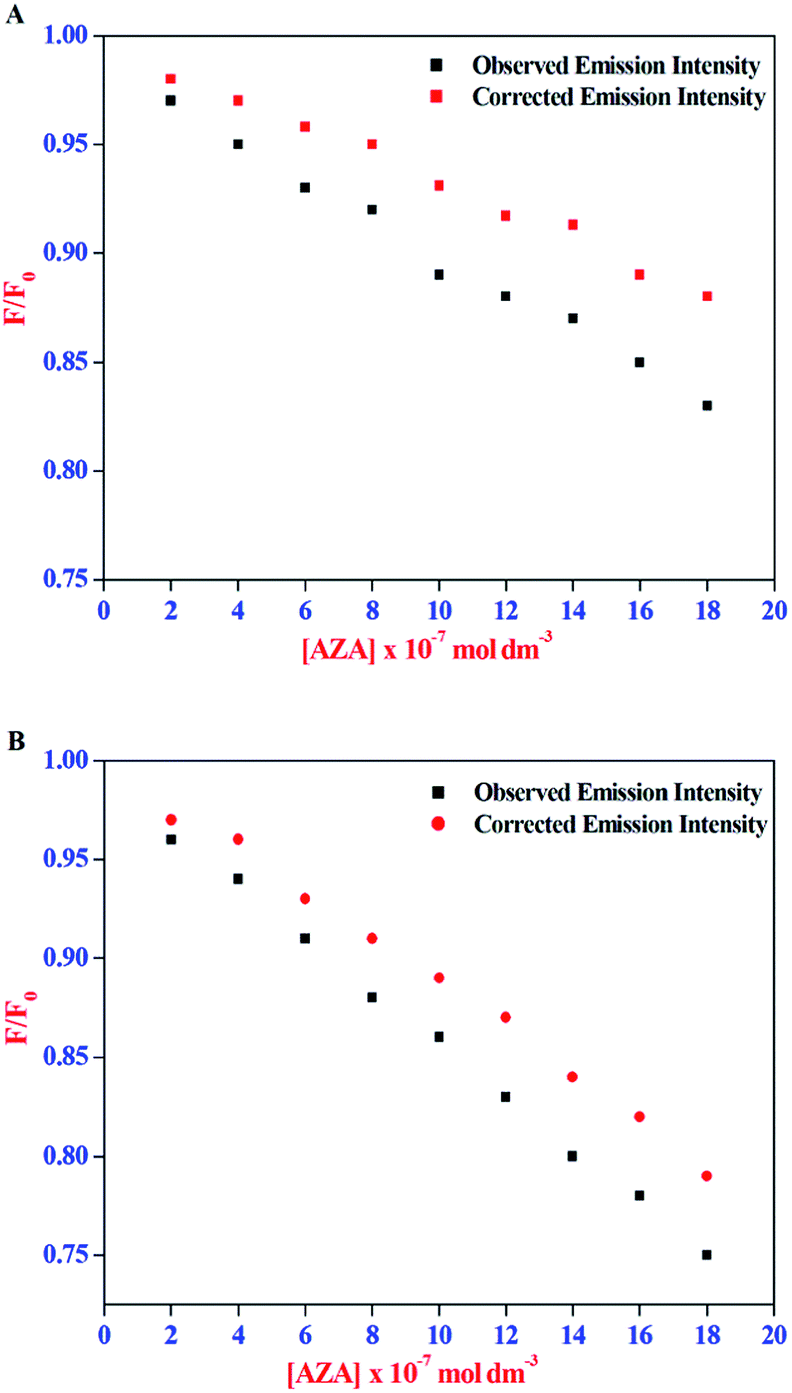
The corrected emission intensity Fcorr was generated from the observed emission intensity Fobs according to eqn (1). The plots of normalized emission (F/F0) of both corrected emission intensities Fcorr and observed emission intensities Fobs in the case of HSA/BSA versus added AZA concentrations are displayed in Fig. 3(A) and (B). The F/F0 values of corrected and observed emission intensities decreased progressively with the increment of AZA concentrations and the former was slower than the latter, suggesting that the emission quenching of HSA/BSA was partly caused by the IFE of AZA. However, the reduction in F/F0 values of corrected emission intensity even after the removal of IFE suggests the possibility of either collisional or binding related quenching of HSA/BSA by AZA. Therefore, the specific quenching mechanism of HSA/BSA in the presence of increasing concentrations of AZA was studied in the ensuing section.
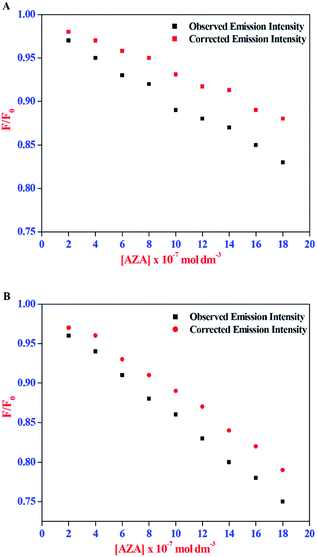 |
| | Fig. 3 Effect of inner-filter effect on the normalized emission intensity (F/F0) of (A) HSA and (B) BSA with the increasing amount of AZA concentration. [AZA]: (a) 0, (b) 2.00 × 10−7, (c) 4.00 × 10−7, (d) 6.00 × 10−7, (e) 8.00 × 10−7, (f) 10.00 × 10−7, (g) 12.00 × 10−7, (h) 14.00 × 10−7 and (i) 16.00 × 10−7 mol dm−3. c[HSA] = c[BSA] = 1.00 × 10−6 mol dm−3. | |
Quenching mechanism of HSA/BSA in presence of AZA
The emission intensity of fluorophores can be decreased by a wide variety of processes. Such decrease in emission intensity is called quenching.28 Quenching can occur by different mechanisms namely collisional and static quenching processes. Collisional quenching occurs when the excited-state fluorophore is deactivated upon contact with some other molecule in solution. In static quenching process, fluorophores form non-fluorescent ground state complexes with quenchers and it does not rely on diffusion or molecular collisions.28 The quenching constant values for the complexation of AZA with HSA/BSA were calculated from the emission spectral data (Fig. 2) using Stern–Volmer equation (eqn (2)).28| |
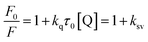 | (2) |
where, F0 and F are the relative corrected emission intensities of HSA/BSA in the absence and presence of quencher, respectively. Kq is the bimolecular quenching rate constant; τ0 is the average lifetime of the fluorophore in the absence of quencher and [Q] is the concentration of the quencher. Ksv is the Stern–Volmer quenching constant which measures the efficiency of quenching.
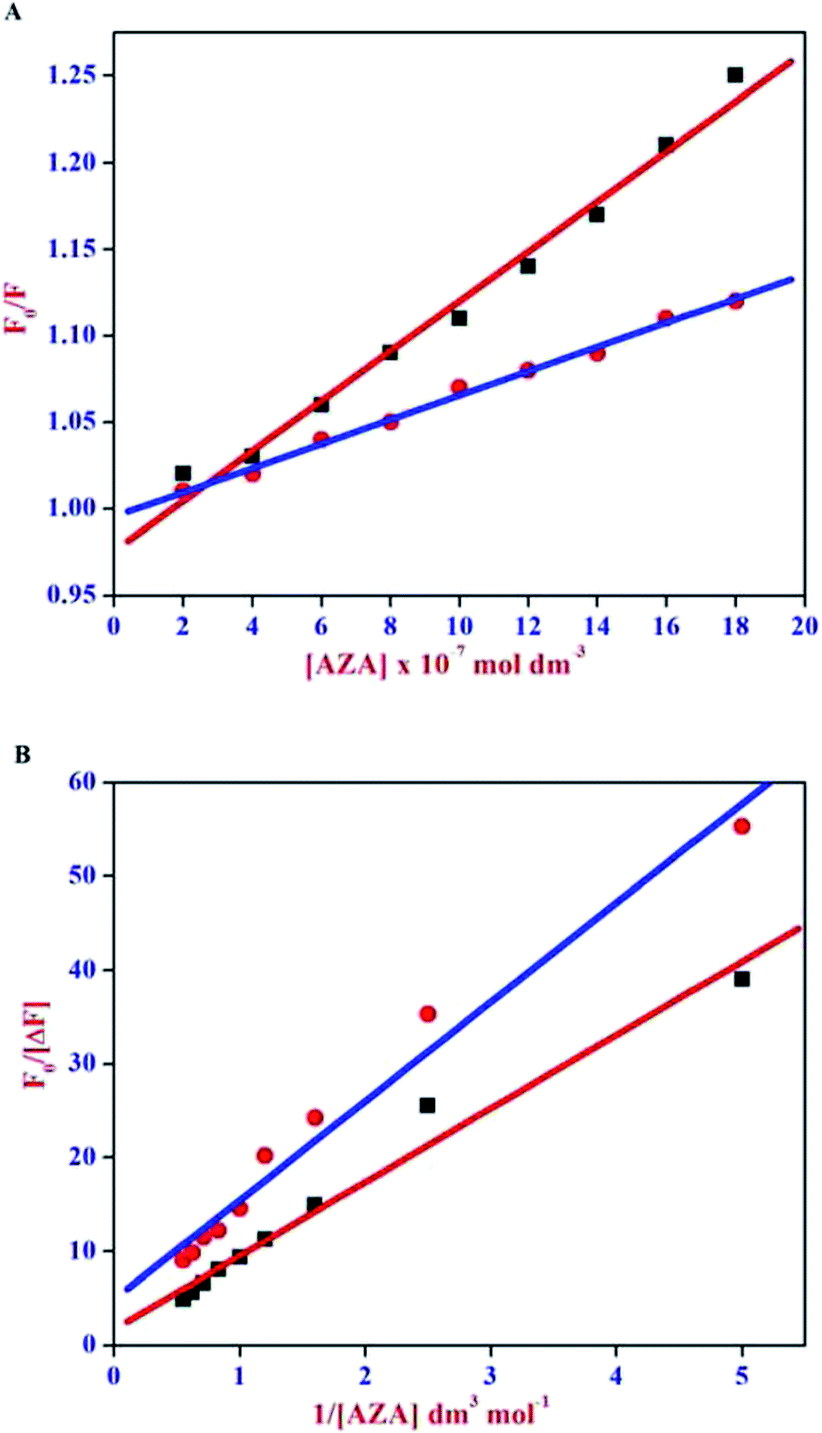
The Stern–Volmer plot for HSA/BSA–AZA system is displayed in Fig. 4(A). As illustrated in Fig. 4(A), the plot of relative corrected emission intensity at maximum wavelength of HSA/BSA vs. the concentration of AZA exhibited a good linear relationship within the investigated concentrations of AZA. The values of Stern–Volmer quenching constant Ksv obtained from the slope and intercept of the linear plot are given in Table 1. The average lifetime (τ0) of a biopolymer has been reported as ∼10−8 s.28 By utilizing the life time (τ0) and Ksv values, the bimolecular quenching constants Kq can be calculated from the relation Kq = Ksv/τ0. The Kq value for the quenching of HSA and BSA by AZA was evaluated as 7.03 × 1012 dm3 mol−1 s−1 and 1.47 × 1013 dm3 mol−1 s−1, respectively. It is well known that for a static quenching process, the quenching constant Kq is far greater than the maximum scatter collision quenching constant (2.00 × 1010 dm3 mol−1 s−1).28 In the present work, the value of Kq for HSA/BSA–AZA system is greater than the limiting diffusion rate constant of the biomolecule, which indicates that the probable quenching mechanism of HSA/BSA in the presence of AZA is initiated by ground state complex formation rather than by dynamic collision.
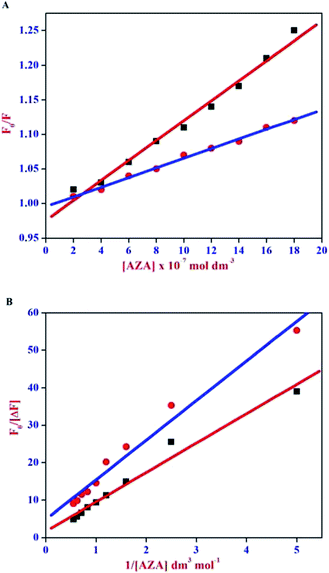 |
| | Fig. 4 (A) Stern–Volmer and (B) Modified Stern–Volmer plot for HSA–AZA [ ] and BSA–AZA [■] complexes. ] and BSA–AZA [■] complexes. | |
Table 1 Binding parameters of HSA–AZA and BSA–AZA complexes
| Binding parameters |
Ksva (dm3 mol−1) |
Kqa (dm3 mol−1 s−1) |
Kaa (dm3 mol−1) |
Ka (dm3 mol−1) |
Kba (dm3 mol−1) |
n |
| The mean value of three individual experiments. |
| HSA–AZA |
(7.03 ± 0.20) × 104 |
(7.03 ± 0.20) × 1012 |
(4.62 ± 0.12) × 105 |
(4.70 ± 0.14) × 105 |
(5.75 ± 0.16) × 105 |
1.12 ± 0.09 |
| BSA–AZA |
(1.47 ± 0.18) × 105 |
(1.47 ± 0.18) × 1013 |
(2.15 ± 0.14) × 105 |
(2.00 ± 0.15) × 105 |
(4.67 ± 0.18) × 105 |
1.09 ± 0.04 |
For a system involving static quenching mechanism, a modified Stern–Volmer equation is applied to calculate the affinity constant (Ka) for the binding of AZA with HSA/BSA. The use of modified Stern–Volmer equation helps in resolving the accessible and inaccessible residues when there is a possibility of differential accessibilities of the protein residues to the probe. The quenching data from the emission spectral studies (Fig. 2) were analyzed according to the modified Stern–Volmer equation (eqn (3)).28
| |
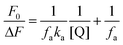 | (3) |
where, Δ
F is the difference in corrected emission intensity of HSA/BSA in the absence and presence of quencher [Q], [(Δ
F =
F0 −
F),
F0 is the corrected emission intensity of HSA/BSA in the absence of AZA and
F is the corrected emission intensity of HSA/BSA at a given concentration of AZA],
fa is the fraction of accessible fluorescence and
Ka is the effective quenching constant for the accessible fluorophores, which is analogous to the associative binding constant for HSA/BSA–AZA system. The plot of
F0/Δ
F vs. 1/[Q] is shown in
Fig. 4(B). As depicted in
Fig. 4(B), the regression curve of
F0/Δ
F against 1/[Q] abides by a linear relationship with a slope equal to the value of (
faKa)
−1. A quantitative estimate of the extent of binding,
i.e., (
Ka), is determined from the intercept to slope ratio of the modified Stern–Volmer equation. The
Ka values for HSA–AZA and BSA–AZA systems are computed as 4.62 × 10
5 dm
3 mol
−1 and 2.15 × 10
5 dm
3 mol
−1, respectively. From the above analysis it is inferred that AZA forms a moderately strong complex with both HSA and BSA. The
Ka values obtained in the present study are consistent with the previous literature reports.
26,27
Binding constant and stoichiometry of HSA/BSA–AZA complexes
The data from emission spectral studies were used to evaluate the binding constant values of AZA with HSA/BSA using the Benesi–Hildebrand equation (eqn (4)).33| |
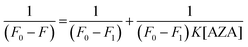 | (4) |
where, F0 is the corrected emission intensity of HSA/BSA in the absence of AZA, F is the corrected emission intensity of HSA/BSA at intermediate concentration of AZA, F1 is the emission intensity of HSA/BSA at infinite concentration of AZA and K is the binding constant.
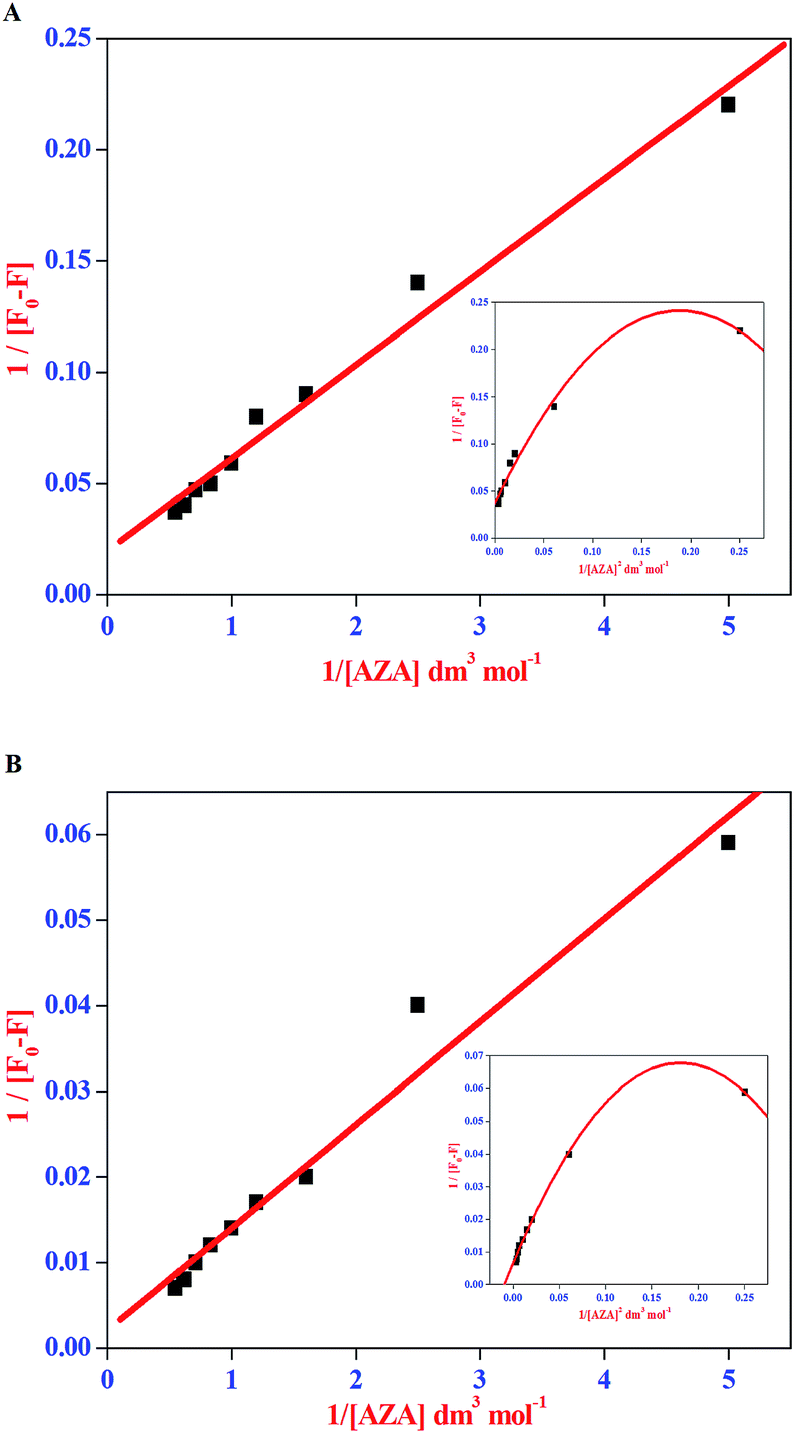
The double reciprocal plots for the complexation of AZA with HSA/BSA are displayed in Fig. 5(A) and (B). It can be seen from Fig. 5(A) and (B), that the plots of 1/[F0 − F] against 1/[AZA] showed a good linearity, thereby suggesting one-to-one interaction between AZA and HSA/BSA. It is widely accepted that the linearity in Benesi–Hildebrand plot is indicative of 1:1 complexation and any deviation from non-linearity is ascribed to the involvement of 1:2 complexation. It has been previously reported by our group, that for a system involving 1:1 complexation process a modified Benesi–Hildebrand plots of 1/[F0 − F] vs. 1/[AZA]2 would result in a non-linear plot.34 Thus, in the present investigation we have applied the modified Benesi–Hildebrand equation to validate the existence of 1:1 complexation and to rule out the possibility of 1:2 complex formation in AZA–HSA/BSA system. As depicted in the inset of Fig. 5(A) and (B) the plots of 1/[F0 − F] against 1/[AZA]2 clearly showed a deviation from linearity. From the above analysis it is inferred that the complexation of AZA with HSA/BSA follows a 1:1 binding stoichiometry. The binding constant values for HSA/BSA–AZA systems are evaluated from the slope and intercept of the linear plots and are summarized in Table 1. The binding constant (K) values summarised in Table 1 falls within the normal range reported earlier for protein–ligand complexation process and it further suggests that the binding of AZA with HSA/BSA is moderately strong.34
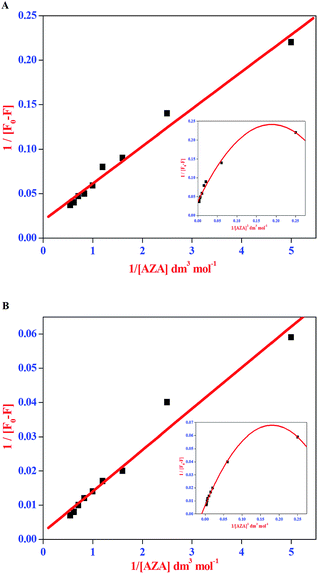 |
| | Fig. 5 Benesi–Hildebrand plot of 1/[F0 − F] vs. 1/[AZA] for binding of AZA with (A) HSA and (B) BSA. Inset shows the modified Benesi–Hildebrand plot of 1/[F0 − F] vs. 1/[AZA]2 for HSA/BSA–AZA systems. | |
The standard free energy changes for the complexation of AZA with HSA/BSA are estimated by using the eqn (5).35
| | |
ΔG = −2.303RTlogK
| (5) |
where, Δ
G is free energy,
K is binding constant at the corresponding temperature (298 K), which can be obtained from the Benesi–Hildebrand plot and
R is the universal gas constant. The free energy change Δ
G involved in the complexation process of HSA–AZA and BSA–AZA system was evaluated as −32.36 kJ mol
−1 and −30.25 kJ mol
−1, respectively. The obtained negative free energy values suggest that the complexation of AZA to HSA/BSA is a spontaneous and highly favourable process and it is consistent with previous reports.
26,34–36
Determination of binding number and binding site on HSA/BSA
The formation of HSA–AZA and BSA–AZA ground state complexes were established by studying the AZA induced quenching of HSA/BSA. In a system involving static quenching process, when small molecules bind independently to a set of equivalent sites on a macromolecule, the binding constant Kb and the number of equivalent binding sites (n) can be calculated by the double logarithmic regression equation (eqn (6)).37| |
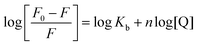 | (6) |
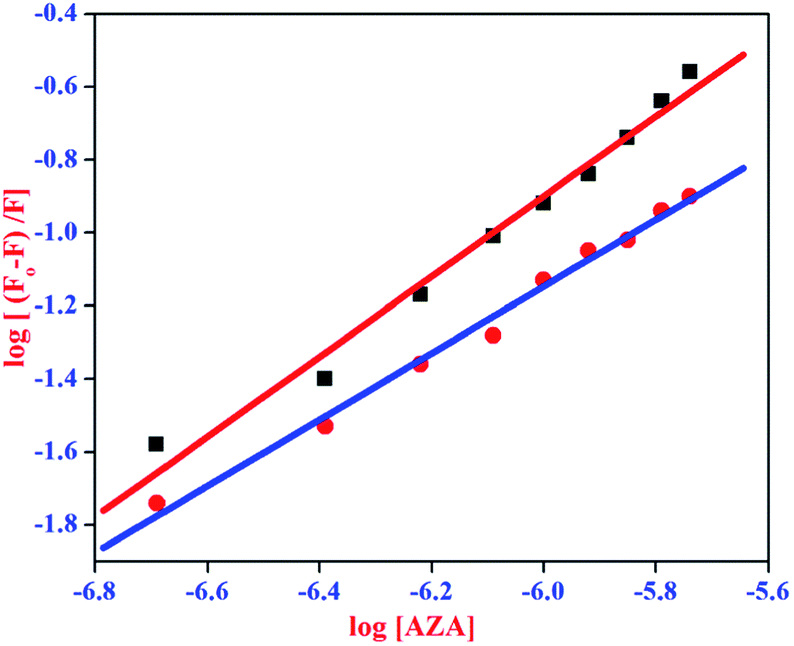
where, F0 and F are same as in eqn (2), n is the average binding number for one AZA molecule and Kb is the binding constant. The double logarithmic plot of log[F0 − F/F] vs. log[AZA] is shown in Fig. 6. As depicted in Fig. 6, the plots of log[(F0 − F)/F] against log[AZA] showed a linear relationship with a slope equal to n. The average binding number for one AZA molecule (n) is approximately equal to 1 for both HSA (1.12) and BSA (1.09). The results thus obtained, indicates the presence of single class of binding site for AZA in both HSA and BSA. Furthermore, the observed values complement the findings of 1:1 binding stoichiometry in the previous section. The calculated binding parameters of HSA–AZA and BSA–AZA complexes are listed in Table 1.
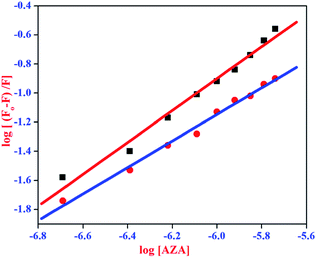 |
| | Fig. 6 Double logarithmic plot for HSA–AZA [ ] and BSA–AZA complexes [■]. ] and BSA–AZA complexes [■]. | |
Site marker-competitive binding experiments
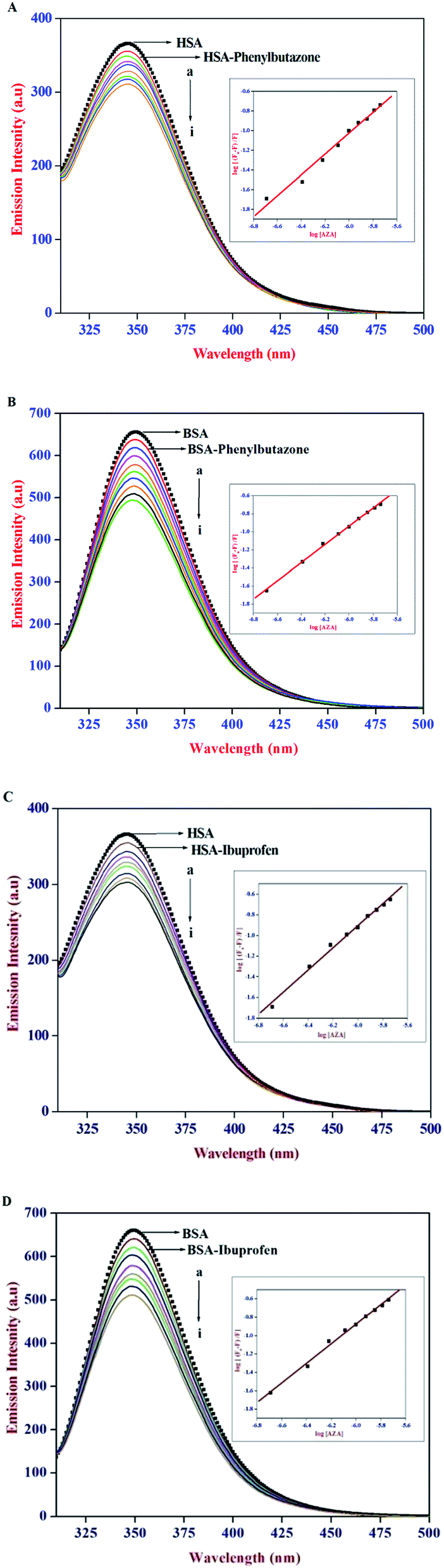
It has been well established that serum proteins are composed of three homologous α-helical domains (I–III) and each domain contains two sub domains. The principal ligand binding sites of HSA/BSA are located in hydrophobic cavities in sub domains IIA and IIIA, which are referred to as site I and site II according to the terminology proposed by Sudlow et al.38 To identify AZA binding site on HSA/BSA, site marker competitive experiments were carried out using the site probes of phenylbutazone for site I and ibuprofen for site II. From the X-ray crystallographic studies, phenylbutazone has been demonstrated to bind to the sub-domain IIA (Sudlow's site I) while ibuprofen is known to be a sub-domain IIIA binder (Sudlow's site II).39,40 Thus, information about the site selective binding interaction of AZA with HSA/BSA can be obtained by monitoring the changes in the emission of AZA bound HSA/BSA in the absence and presence of the two markers separately.
In the site marker competitive experiments, AZA was gradually added to the solution of HSA/BSA and site markers (1.00 × 10−6 mol dm−3) held in equimolar concentrations. As displayed in Fig. 7 the emission spectrum of HSA/BSA (1.00 × 10−6 mol dm−3) in the absence of phenylbutazone exhibits an emission maximum at 346 nm. Upon the addition of equimolar concentration of phenylbutazone (1.00 × 10−6 mol dm−3) to the HSA/BSA solution, the emission intensity of HSA/BSA showed a slight decrease and it is depicted in Fig. 7(A) and (B). These observations implies that phenylbutazone has been bound to HSA/BSA. Increasing concentration of AZA was added to the equimolar solution of HSA/BSA–phenylbutazone and the resulting spectra are displayed in Fig. 7(A) and (B). If AZA binds to HSA/BSA in the same site as that of phenylbutazone it has to compete with phenylbutazone in binding to HSA/BSA. As displayed in Fig. 7(A)(a–i) and 7B(a–i) the addition of varying concentrations of AZA decreased the emission intensity of HSA/BSA–phenylbutazone system gradually decreased and the resulting quenching data were utilized to evaluate the binding constant value. The comparison of emission spectra of HSA/BSA in the absence and presence of ibuprofen displayed in Fig. 7(C) and (D), clearly suggested the binding of ibuprofen to HSA/BSA. It can be inferred from Fig. 7(C) and (D), that the titration of AZA with HSA/BSA solution held in equilibrium concentration of ibuprofen resulted in the modification of emission profiles of both HSA and BSA. The emission quenching data from the site marker experiments were analyzed according to the double logarithmic eqn (6). The double logarithmic plots for HSA/BSA in the presence of phenylbutazone and ibuprofen are presented in the inset of Fig. 7(A)–(D), respectively. The binding constant Kb values for HSA/BSA–AZA system in the presence of site markers were estimated to compare the influence of phenylbutazone and ibuprofen on the binding of AZA to HSA/BSA. The computed values from the double logarithmic plot are listed in Table 2. As depicted in the inset of Fig. 7(A)–(D), the plot of eqn (6) complies with a linear regression, indicating the existence of possible competition between AZA and phenylbutazone or ibuprofen in binding to HSA/BSA. It can be noticed from Table 2 that the binding constant values in presence of phenylbutazone is much smaller than that of the binding constant values in presence of ibuprofen for both HSA and BSA. Moreover, the binding constant value in presence of ibuprofen showed a very small deviation from that of HSA/BSA–AZA systems. From the above interpretation, it is revealed that AZA mainly competes with phenylbutazone in sub domain IIA (Sudlow's site I) and it binds in site I with high affinity. It has been recently reported by our group that a structurally similar phenothiazine dye Toluidine Blue O binds to site I of both HSA and BSA.26 Phenothiazine dyes are known to undergo 1:1 complexation with β-cyclodextrin via hydrophobic interactions.41 Therefore, in the present case it is believed that AZA could bind to site I of HSA/BSA driven by hydrophobic interaction. However, the role of other possible interactions such as electrostatic, hydrogen bonds, van der Waals interactions and steric contacts cannot be ruled out in the binding of AZA to HSA/BSA. Moreover, it is well known that serum albumins possess a net negative charge at physiological pH and AZA carries a positive charge in aqueous solution.38 Hence, the binding of AZA to HSA/BSA might involve electrostatic interactions as well.
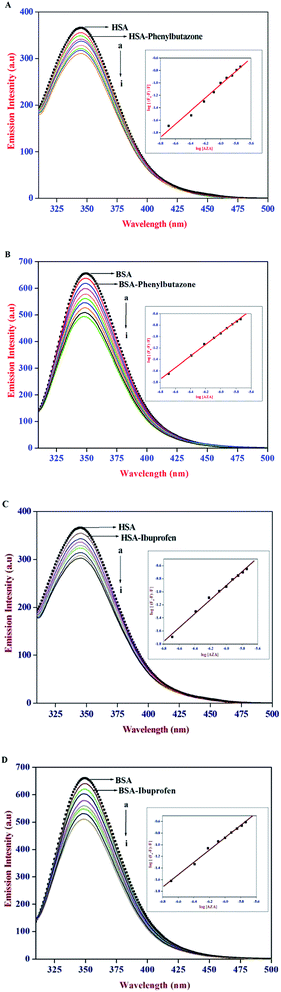 |
| | Fig. 7 Emission spectra of (A) HSA–phenylbutazone, (B) BSA–phenylbutazone, (C) HSA–ibuprofen and (D) BSA–ibuprofen [1:1] at various AZA concentrations. [AZA]: (a) 0, (b) 2.00 × 10−7, (c) 4.00 × 10−7, (d) 6.00 × 10−7, (e) 8.00 × 10−7, (f) 10.00 × 10−7, (g) 12.00 × 10−7, (h) 14.00 × 10−7 and (i) 16.00 × 10−7 mol dm−3. Inset shows the respective double logarithmic plot. | |
Table 2 Estimated binding constants for site marker competitive experiments of HSA–AZA and BSA–AZA systems
| Site marker |
Binding constant Kba (dm3 mol−1) for HSA |
Rb |
Binding constant Kba (dm3 mol−1) for BSA |
Rb |
| The mean value of three individual experiments. R is the correlation coefficient. |
| Blank |
(5.75 ± 0.16) × 105 |
0.99 |
(4.67 ± 0.18) × 105 |
0.99 |
| Phenylbutazone |
(2.13 ± 0.14) × 105 |
0.99 |
(1.31 ± 0.15) × 105 |
0.98 |
| Ibuprofen |
(4.50 ± 0.13) × 105 |
0.98 |
(3.16 ± 0.12) × 105 |
0.99 |
Red-edge excitation shift of HSA/BSA in the presence of AZA
Red edge excitation shift (REES) is a shift in the emission maximum towards a higher wavelength caused by a shift in the excitation wavelength towards the red edge of the absorption band. REES occurs due to the electronic coupling between Trp indole rings and neighbouring dipoles and occurs when there are slow relaxations of solvent media.28 REES of fluorescence can be used as a parameter for studying the photophysical and chemical properties of isolated proteins.43 This phenomenon is an indication for a strongly reduced dynamic environment of a single Trp, which has a very low accessibility to the solvent and is caused by electronic coupling between Trp indole rings and neighbouring dipoles. Thus, REES is particularly useful in monitoring motions around the Trp residues in protein study.42 In the present investigation, we chose to excite the Trp at both 295 and 310 nm to investigate the REES effect, and the results are listed in Table 3. The value of Δλem-max is defined as the difference of the emission maximum obtained for the excitation wavelength at 295 nm and 310 nm, respectively. As shown in Table 3, native HSA/BSA showed 6 nm and 2 nm REES, respectively, indicating that Trp residues in albumin were in a slight motionally restricted environment. In the presence of AZA, the REES values of both HSA and BSA showed a decrease indicating that the addition of AZA had an obvious impact on the mobility of the Trp microenvironment with the motional restriction of Trp residues relaxed to an extent. Similar kind of reverse REES trends have been reported in the case of Ag NPs interaction with BSA.43
Table 3 Red edge excitation shift effects at λex 295 nm and λex 310 nm
| Sample |
λem-max (nm) |
Δλem-max (REES) (nm) |
| λex 295 nm |
λex 310 nm |
| HSA |
348 |
354 |
6 |
| HSA–AZA |
348 |
353 |
5 |
| BSA |
349 |
351 |
2 |
| BSA–AZA |
350 |
351 |
1 |
Conformational studies of HSA/BSA
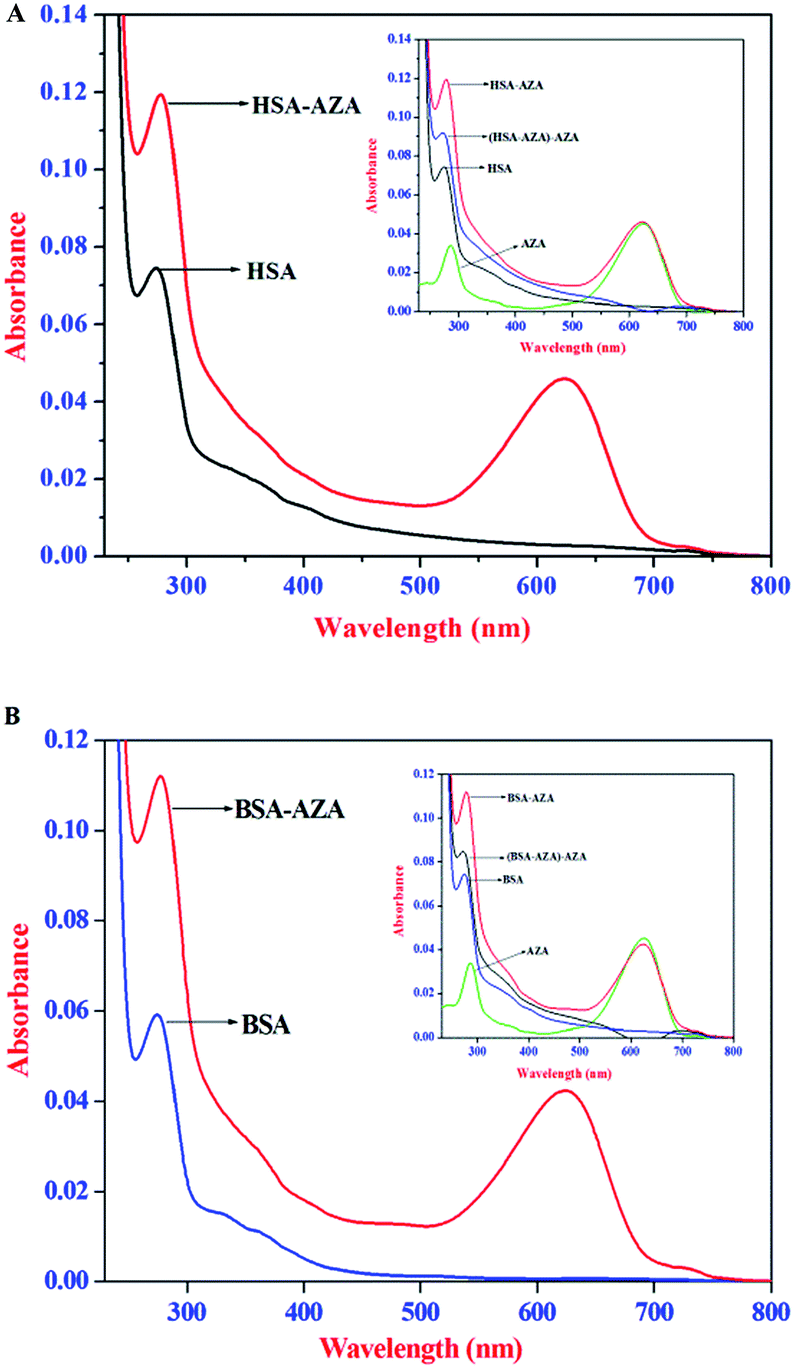
Absorption spectral studies of HSA/BSA with AZA
UV-Vis absorption spectroscopy is an ideal tool to explore the structural changes of protein and to investigate protein–ligand complex formation. HSA/BSA exhibit an absorption maximum at 278 nm which mainly originates from the aromatic amino acid residues, viz., Trp, Tyr, and Phe.26 It is well known that the absorption maximum of HSA/BSA is highly sensitive to the surrounding microenvironment and it displays a substantial spectral shift upon changes in protein conformation.26,38 Consequently, in the present study, we have carried out absorption spectral studies of HSA/BSA and AZA at equimolar concentrations [1:1] to explore AZA induced structural changes in serum albumins and the resulting spectra are displayed in Fig. 8(A) and (B). It is pertinent to note from Fig. 8(A) and (B), that upon equimolar addition of AZA, the absorption intensity of HSA/BSA at 278 nm showed an increase with a simultaneous red shift in the absorption maximum from 278 nm to 283 nm. It is widely emphasized that the interaction between serum proteins and ligand molecules often result in perturbation of protein absorption spectrum.38 In recent past, similar kind of absorption spectral changes have been attributed to alterations in the micro-environment of Trp and Tyr residues of BSA.44 The hyperchromism at 278 nm clearly shows that more aromatic acid residues were extended into the aqueous environment. The Trp residue, which was originally buried in a hydrophobic pocket, was exposed to an aqueous milieu to a certain degree.44 The results from absorption spectral measurements enabled us to conclude that AZA has induced tertiary structural changes in HSA/BSA and considerable microenvironmental changes in the vicinity of Trp and Tyr residues.44 As shown in the inset of Fig. 8(A) and (B), the absorption spectra of HSA/BSA and the difference absorption spectra of HSA/BSA:AZA [1:1] complex and AZA could not be superposed within the experimental error. This observation is found to bear good consistency with previous literature reports for ground state complexation of various drugs with protein.26,38 It is widely emphasized that the formation of ground state complex between the protein and ligand, often leads to alteration in the absorption spectrum of the proteins.28,38 Subsequently, the perturbation in the absorption spectra of serum proteins suggests that the quenching of HSA/BSA by AZA is primarily initiated by static quenching process and this corroborates well with the results obtained from the emission spectral studies.
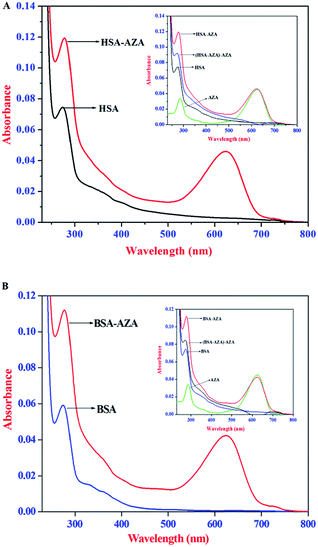 |
| | Fig. 8 Absorption spectra of (A) HSA alone and HSA–AZA complex; (B) BSA alone and BSA–AZA complex. Conditions: c[HSA] = c[BSA] = 1.00 × 10−6 mol dm−3; c[AZA] = 1.00 × 10−6 mol dm−3. Inset shows the difference absorption spectra of HSA/BSA:AZA [1:1] complex and AZA. | |
Constant wavelength synchronous emission spectroscopy of HSA/BSA–AZA complexes
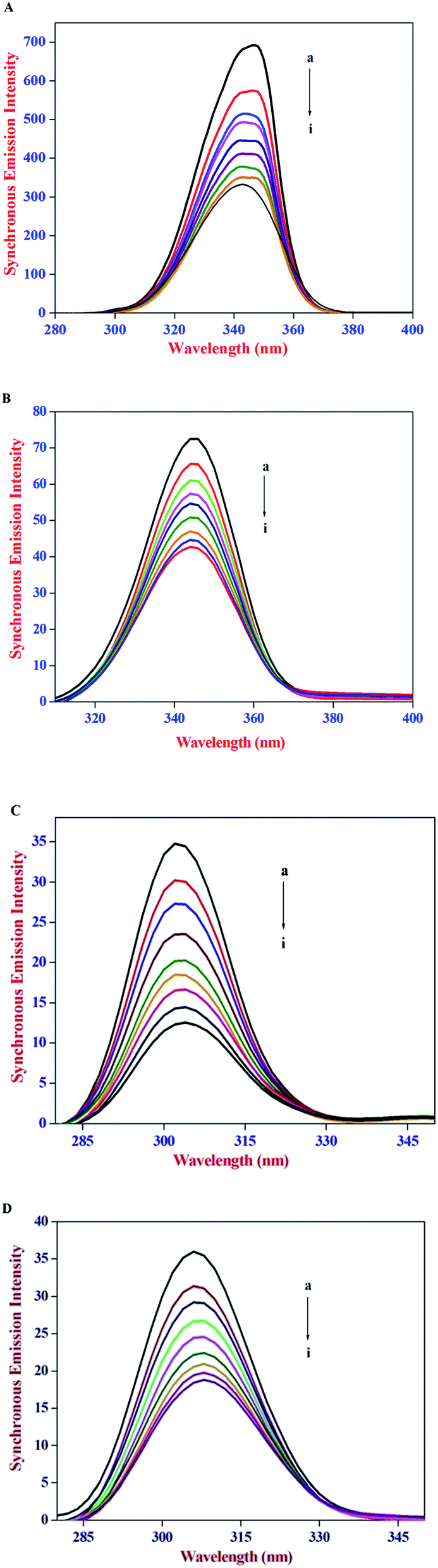
The constant wavelength synchronous emission spectroscopy technique introduced by Lloyd45,46 involves simultaneous scanning of the excitation and the emission monochromators while maintaining a constant wavelength interval between them. Constant wavelength synchronous emission spectroscopy has been used to characterize complex mixtures providing finger prints of complex samples and it gives information on the molecular environment in the vicinity of Tyr and Trp residues and has several advantages, such as sensitivity, spectral simplification, spectral bandwidth reduction and avoiding different perturbing effects. According to Miller's theory,47 when Δλ between excitation and emission wavelength is stabilized at 15 nm or 60 nm, the spectrum characteristic of the protein Tyr or Trp residues was observed. Subsequently, Yuan et al.48 suggested a useful method to study the environment of amino acid residues in proteins by measuring the possible shift in emission maximum λemi. In synchronous emission spectra, the decrease in emission intensity without any shift indicates that the microenvironment around that particular residue is not disturbed. In contrast, a red-shift is indicative of an increase in the hydrophilicity around the fluorophore in serum albumin and blue-shift is attributed to an increase in the hydrophobicity around the fluorophore moiety.49
The constant wavelength synchronous emission spectra of HSA/BSA in the presence of increasing concentrations of AZA are shown in Fig. 9(A)–(D). As can be seen from Fig. 9(A) and (B), the maximum emission of Trp in HSA has a slight blue shift from 344 nm to 341 nm, whereas in the case of BSA a similar blue shift of Trp emission wavelength from 349 nm to 344 nm was observed. As depicted in Fig. 9(C) and (D) the characteristic emission spectrum of Tyr has a slight red shift from 301 nm to 303 nm for HSA and 304 nm to 307 nm for BSA. The above results indicated that in the presence of AZA the conformations of both HSA and BSA changed and the polarity around the Trp residues decreased with increase in hydrophobicity.50 It is apparent from the above analysis that the synchronous emission intensity of both Trp and Tyr decreased instantaneously upon the addition of AZA dye and this corroborates the occurrence of emission quenching in the binding process. The results from synchronous emission study further suggests that the binding regions of AZA are located in the vicinity of both Trp and Tyr residues of HSA/BSA, since a distant event cannot cause the synchronous emission quenching of both Trp and Tyr residues. Therefore, from constant wavelength synchronous emission experiments it can be concluded that the binding site of AZA in HSA/BSA is most probably located adjacent to Trp or Tyr residues.
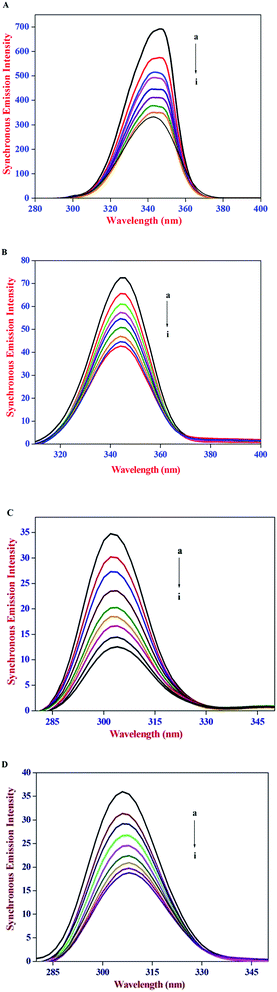 |
| | Fig. 9 Synchronous emission spectrum of (A) HSA at Δλ = 60 nm, (B) BSA at Δλ = 60 nm, (C) HSA at Δλ = 15 nm and (D) BSA at Δλ = 15 nm at various AZA concentrations. | |
Circular dichroism spectroscopy of HSA/BSA–AZA complexes
Circular dichroism (CD) spectroscopy is being increasingly recognized as a valuable technique for examining the structure of proteins in solution. CD signals arise where absorption of radiation occurs and thus spectral bands are easily assigned to distinct structural features of a molecule. An advantage of the CD technique in studies of proteins is that complementary structural information can be obtained from a number of spectral regions. It is possible to estimate the α-helical content of proteins by using the values of CD signals at 208 nm and 222 nm. To gain better understanding on the AZA induced conformational changes in HSA/BSA, the CD spectra of HSA/BSA in the absence and presence of AZA was obtained. The α-helical content of HSA/BSA was evaluated from eqn (7) and eqn (8).51| |
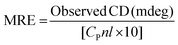 | (7) |
| |
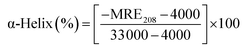 | (8) |
where, MRE is the mean residual elipticity, CP is the molar concentration of the protein, n is the number of amino acid residues (583 amino acids for BSA and 585 amino acids for HSA), l is the path length of the cell, MRE208 is the observed MRE value at 208 nm, 4000 is the MRE of the β-form and random coil conformation at 208 nm and 33000 is the MRE value of a pure α-helix at 208 nm.
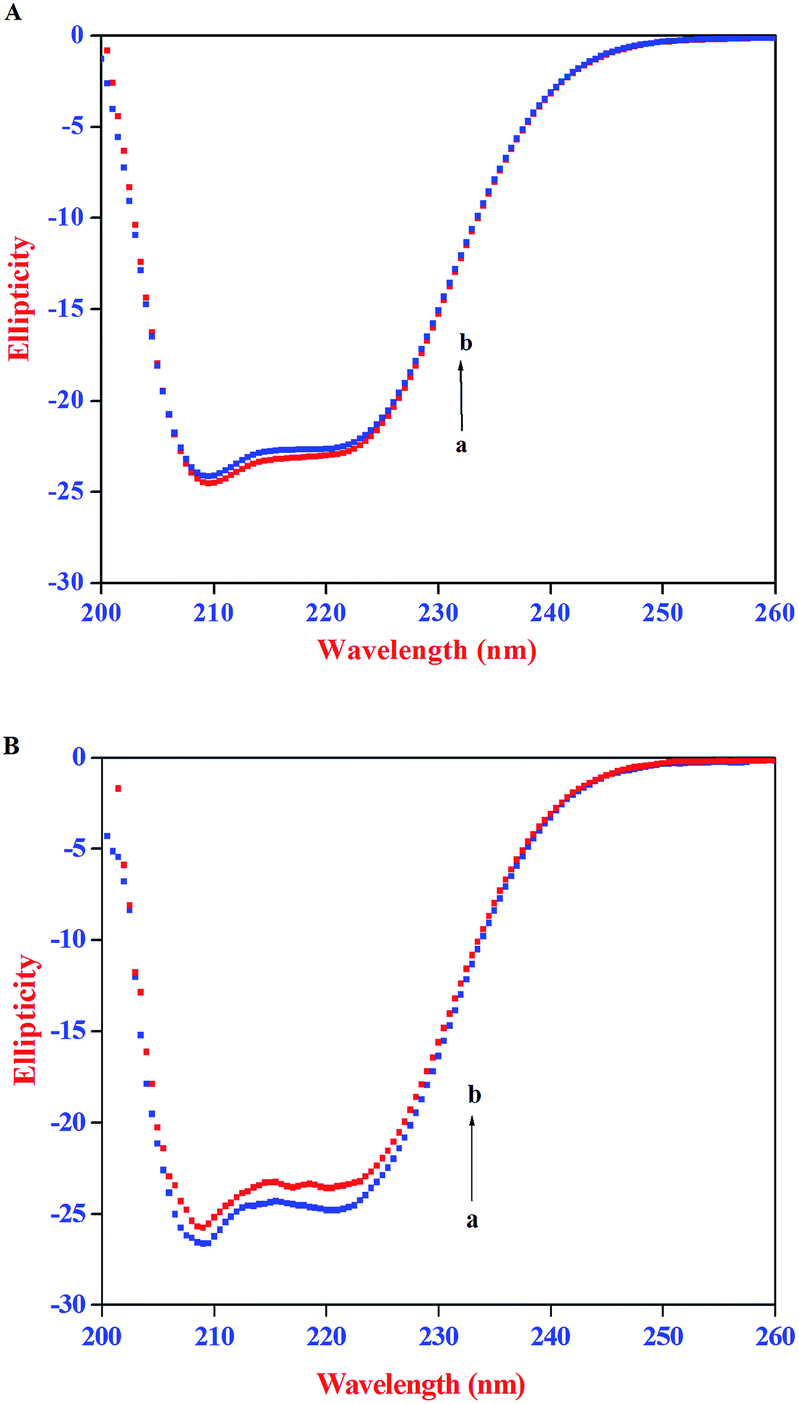
The CD spectra of HSA/BSA in the absence and presence of AZA is shown in Fig. 10. As shown in Fig. 10(A) and (B), the CD spectrum of HSA/BSA, in the absence of AZA exhibits negative absorption bands with maxima at 222 nm (n–π*) and 208 nm (π–π*), which are characteristic of α-helical structure of protein. Upon the addition of AZA, the CD spectrum of HSA/BSA (Fig. 10(A) and (B)) showed a decrease in band intensity of both the negative bands at 208 nm and 220 nm without any significant shift of the peaks, indicating that AZA dye induced a slight decrease in the α-helical content of HSA/BSA. Similar kind of behaviour has been reported in the case of 1-phenylisatin binding to BSA.52 By utilizing the eqn (7) and (8), quantitative analyses of the α-helical content were obtained and are listed in Table 4. It was deduced that α-helical content in both HSA and BSA showed a decrease upon the addition of AZA. From the CD spectral analysis, it is concluded that the interaction of AZA with HSA/BSA caused a change in the secondary structure of protein, with the loss of helical stability.
 |
| | Fig. 10 CD spectra of (A) HSA and (B) BSA in the absence and presence of AZA. Conditions: [HSA] = [BSA] = 2.00 × 10−6 mol dm−3; [AZA]: (a) 0 × 10−6 mol dm−3 and (b) 2.00 × 10−6 mol dm−3. | |
Table 4 Percentage α-helical content of HSA/BSA in the absence and presence of AZA
| System |
% α-Helix content |
| HSA |
BSA |
| Serum albumins |
56.94 ± 2 |
64.01 ± 2 |
| Serum albumins + AZA |
54.96 ± 2 |
61.29 ± 2 |
Three dimensional (3D) emission spectral studies of HSA/BSA with AZA
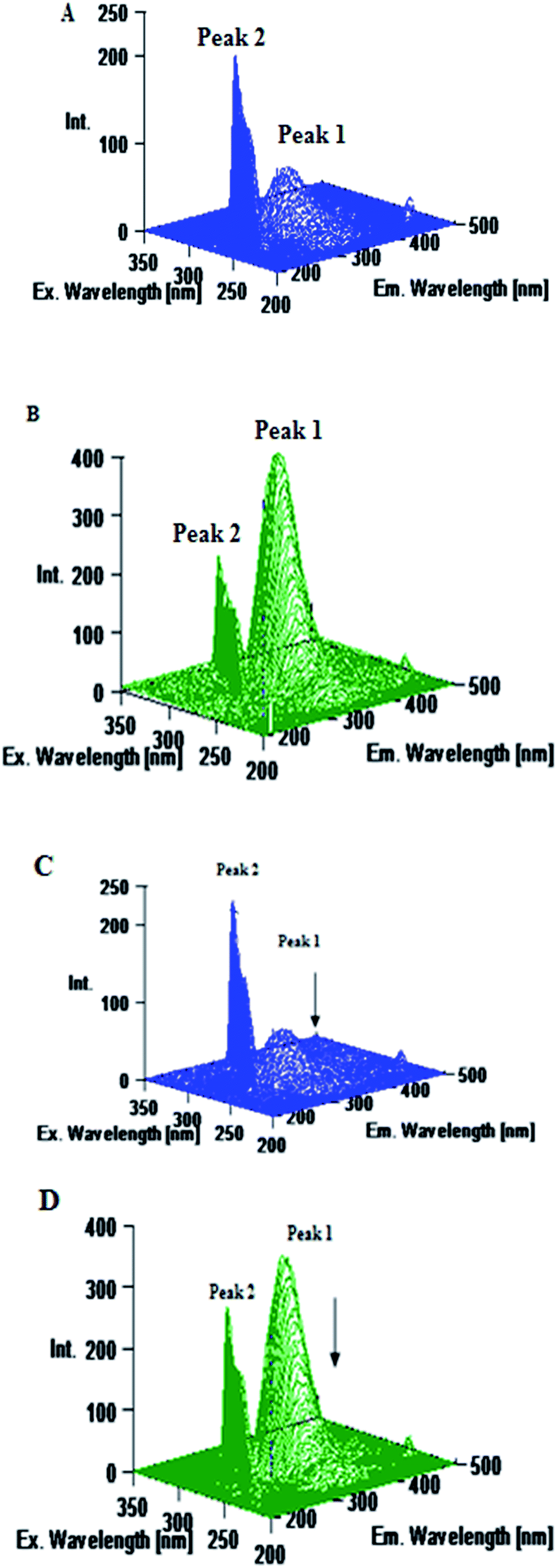
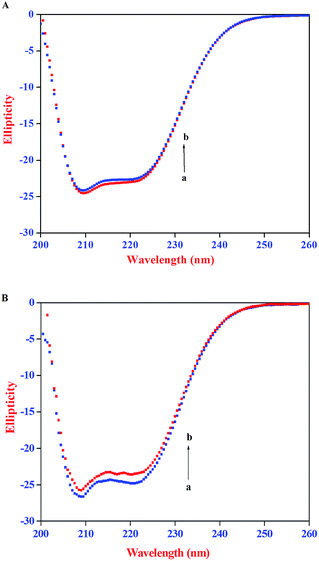
Excitation–emission matrix spectroscopy or 3D emission spectroscopy can provide total information regarding the emission characteristics of fluorophores by changing the excitation and emission wavelength simultaneously. Accordingly, 3D emission spectroscopy can be employed to yield information regarding the conformational change of protein bound to a probe. It is well established that the 3D emission spectral analysis of Trp excitation and emission spectra can provide complete information about the changes in protein conformations.26,53 The AZA induced conformational and microenvironmental changes of HSA/BSA were investigated by 3D emission spectroscopy. The 3D emission spectra of HSA/BSA in the absence and presence of AZA are displayed in Fig. 11. From Fig. 11(A) and (B) it can be noticed that the 3D emission spectrum of HSA/BSA shows two peaks namely peak 1 and peak 2. Peak 1 is the Rayleigh scattering peak (λem = λex) and peak 2 reveals the spectral behaviour of Trp and Tyr residues of HSA/BSA and it is closely related to microenvironmental polarity around Trp and Tyr residues.26,53,54 As depicted in Fig. 11(C) and (D) the emission intensity of peak 1 decreases upon the addition of AZA (1.0 × 10−6 mol dm−3) to HSA/BSA. The observed changes in the emission intensity of peak 1 and peak 2 indicate that the interaction of AZA with HSA/BSA induced a change in the secondary structure of both HSA and BSA. From the above spectral analyses, it can be concluded that the interaction of AZA with HSA/BSA induces a slight conformational change in the protein structure which complies well with the absorption and CD spectral measurements.
 |
| | Fig. 11 Three dimensional emission spectra of (A) HSA alone, (B) BSA alone, (C) HSA–AZA complex and (D) BSA–AZA complex. Conditions: c[HSA] = c[BSA] = 1.00 × 10−6 mol dm−3; c[AZA] = 1.00 × 10−6 mol dm−3. | |
Molecular docking studies of HSA/BSA–AZA complexes
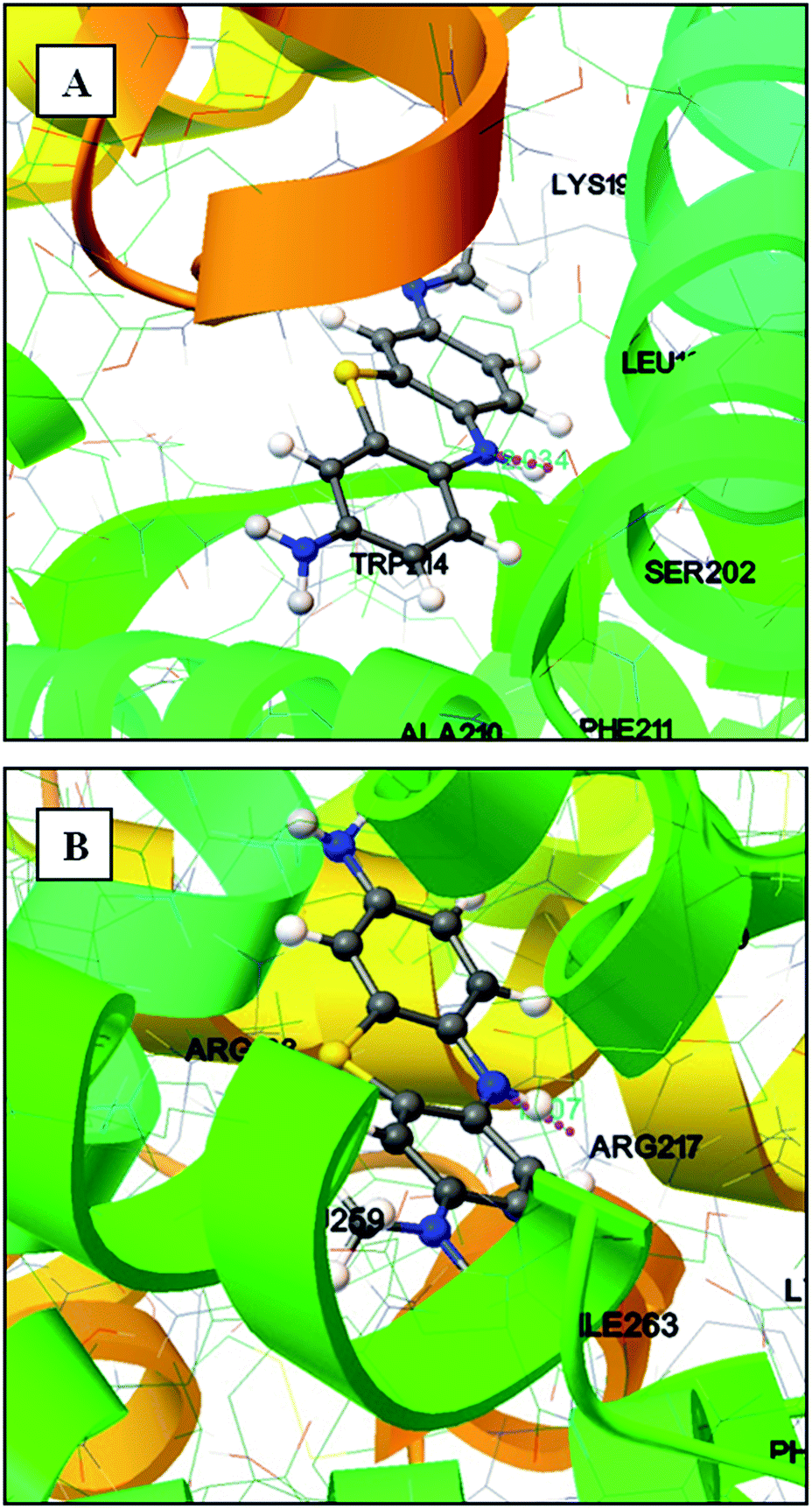
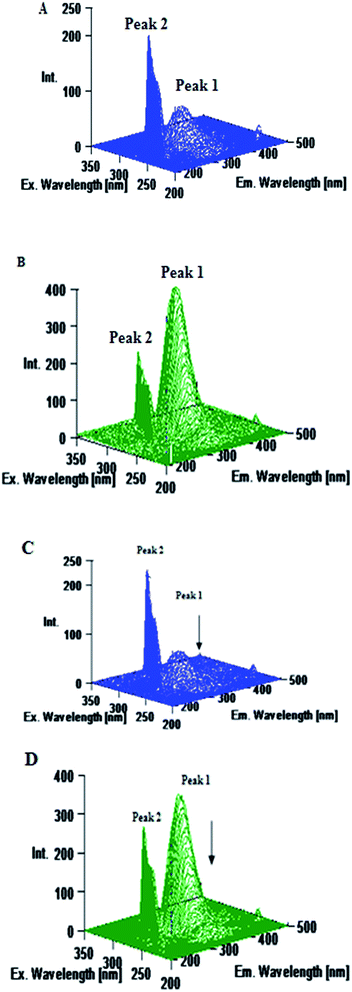
To understand the efficacy of a photoactive dye molecule, the knowledge of its binding location in the protein environment is very crucial and important.55 In the present investigation, AutoDock-based blind docking has been employed as the actuating strategy to substantiate the site competitive experimental results. The results from site competitive experiments revealed the probable binding location of AZA as site I of HSA/BSA. Therefore, the whole region of subdomain II A of HSA/BSA was extracted for docking with AutoDock.56 During the docking process, the lowest binding energy conformer was searched out of 25 different conformers for each docking simulation and the resultant one was used for further analysis. As is usual in a blind docking simulation protocol, we obtained a number of binding sites and the best ranked result, which has the lowest free energy for HSA/BSA are shown in Fig. 12(A) and (B). The docking summary of HSA/BSA–AZA complexes listed in Table 5, provide a convincing evidence for probable binding location of AZA in subdomain II A (Sudlow's site I) of HSA/BSA. The docked pose of AZA–HSA/BSA complexes displayed in Fig. 12(A) and (B) reveals subdomain II A of HSA/BSA to be the favorable binding site for AZA. It has been well established that in HSA, the inside wall of the pocket of subdomain IIA is formed by hydrophobic side chains, whereas the entrance of the pocket is surrounded by positively charged residues consisting of Arg-257, Arg-222, Lys-199, His-242, Arg-218 and Lys-195.57 It can be seen from Fig. 12(A) that the AZA molecule was penetrating deep inside the binding pocket and it is located adjacent to amino acid residues Lys-199, Leu-198, Ala-210 and Trp-214 of subdomain IIA of HSA. In addition, HG atom (G indicates the position of H atom in AZA) of AZA forms a hydrogen bond with Ser-202 with a bond length of 2.034 Å. The calculated free energy change for HSA–AZA complex was computed as −5.05 kcal mol−1. In the case of BSA, the amino acid residues in subdomain IIA are comprised of Trp-213, His-237, Leu-241, Tyr-149, Arg-198, Gln-195, Arg-194, Ala-290, Glu-291 and Arg-217.57 These amino acid residues essentially undergoes interaction via hydrophobic and van der Waals forces. However, in certain cases these amino acid residues undergo hydrogen bonding and hydrophobic interactions as well.24,26 In the docking study of BSA–AZA system, we observed a formation of one hydrogen bond with a bond length of 1.807 Å between Arg-217 and HE atom (E indicates the position of H atom in AZA) of AZA with a free energy value of −5.75 kcal mol−1 (Fig. 12(B)). The outcome of the docking studies suggested that the formation of hydrogen bonds between AZA and HSA/BSA could have resulted in the decrease of hydrophilicity in the binding cavity. As a result of it, the hydrophobicity within the active site of the proteins is increased, which in turn leads to the stabilization of HSA/BSA–AZA complexes.
 |
| | Fig. 12 (A) Molecular docking of HSA–AZA complex. (B) Molecular docking of BSA–AZA complex. Hydrogen bond is shown in pink dotted line. | |
Table 5 Docking summary of HSA and BSA with AZA
| Rank |
Run |
Binding energy (kcal mol−1) |
Estimated inhibition constant (Ki) |
| HSA |
BSA |
HSA |
BSA |
HSA |
BSA |
| 1 |
7 |
6 |
−5.05 |
−5.75 |
200.39 μM |
60.64 μM |
| 2 |
15 |
14 |
−5.04 |
−5.75 |
201.91 μM |
60.67 μM |
| 3 |
13 |
5 |
−5.04 |
−5.75 |
202.94 μM |
60.69 μM |
| 4 |
17 |
3 |
−5.03 |
−5.75 |
205.13 μM |
60.73 μM |
| 5 |
1 |
4 |
−5.03 |
−5.75 |
205.19 μM |
60.74 μM |
The free energy change (ΔG) and binding constant (Kb) values estimated from the computational studies for HSA–AZA system were found to be −21.12 kJ mol−1 and 0.49 × 104 dm3 mol−1 respectively. In the case of BSA–AZA system, the values obtained for ΔG and Kb was −24.05 kJ mol−1 and 1.64 × 104 dm3 mol−1 respectively. The ΔG and Kb values evaluated from computational studies showed a deviation from that of the experimentally calculated values for both HSA–AZA (ΔG = −32.36 kJ mol−1 and Kb = 4.70 × 105 dm3 mol−1) and BSA–AZA (ΔG = −30.25 kJ mol−1 and Kb = 2.00 × 105 dm3 mol−1) systems. The difference in computational and experimental values can be explained based on the fact that X-ray structure of the protein from crystals is different from that of the aqueous system used in the study, which results in the difference of the microenvironment around the ligand.25 Similar kind of results were observed in coumaroyl tyramine and naringin binding to HSA.26,57,58 However, slight inadequacy in force fields and algorithms of docking may also contribute to the deviation in computed free energy values from that of the experimental results. Thus, from the docking simulation studies it is inferred that the existence of hydrophobic interaction in combination with hydrogen bonding and electrostatic interactions played a vital role in AZA binding to site I of HSA/BSA. It is pertinent to note from the docking results that AZA molecule is located in the vicinity of Trp-214 residue of HSA and Trp-213 residue of BSA. This observation further substantiates the findings of emission titration experiments of HSA/BSA–AZA complexes. Thus, the molecular docking studies of HSA/BSA–AZA complexes have provided a good structural basis to correlate the results with that of the emission quenching studies of HSA/BSA in the presence of AZA.
Conclusion
In conclusion, the results discussed herein elucidate the influence of inner filter effect on the emission quenching of HSA/BSA even at low concentration of AZA. The emission titration experiments between AZA and HSA/BSA demonstrated the existence of static quenching mechanism. The calculated binding constant values suggested that the complexation process of AZA with HSA/BSA is characterized by reasonable binding constant values. The estimated negative free energy change values revealed that the binding of AZA to HSA/BSA occurs spontaneously. The site marker competitive experiments indicate that AZA binds specifically to the hydrophobic pocket of site I (sub domain IIA) of HSA/BSA. The results from molecular docking studies reiterate site I as the favourable binding site for AZA. Constant wavelength synchronous emission and 3D emission spectral analysis revealed that the polarity around the Trp residue was decreased and the secondary structural conformation of HSA/BSA was altered due to the binding of AZA. The AZA induced conformational changes of HSA/BSA was further established using absorption and CD spectral studies. The present study could provide accurate and comprehensive information pertaining to the binding mechanism of AZA with HSA/BSA. This in vitro knowledge could help in understanding the effect of AZA on protein function during its transportation and distribution in blood.
Acknowledgements
The authors' gratefully acknowledges Department of Science and Technology (DST-SERC-FAST Track scheme, Project no. SR/FT/CS-015/2009) and University Grants Commission (UGC-MRP, Project no. 41-309/2012 (SR)), India for the financial support. Prof. A. Ramu and Prof. R. Ramaraj of Madurai Kamaraj University are gratefully acknowledged for their help in recording the CD spectra.
References and Notes
- L. M. Moreira, J. P. Lyon, A. P. Romani, D. Severino, M. R. Rodrigues and H. P. M de Oliveira, Phenotiazinium dyes as photosensitizers (PS) in photodynamic therapy (PDT): spectroscopic properties and photochemical mechanisms, in Advanced aspects of spectroscopy, 2012, ch. 14, pp. 393–422, DOI:10.5772/48087.
- J. Rojo, S. M. Picker, J. J. G. García and B. S. Gathof, Rev. Med. Hosp. Gen., 2006, 69, 99–107 Search PubMed.
- J. L. Vennerstrom, M. T. Makler, C. K. Angerhofer and J. A. Williams, Antimicrob. Agents Chemother., 1995, 39, 2671–2677 CrossRef CAS.
- S. M. Weiss, PCT Int. Appl. WO 2007022568, Chem. Abstr., 2007, vol. 146, p. 280994.
- P. B. Weisz, PCT Int. Appl. WO 9711090, Chem. Abstr., 1997, vol. 126, p. 272367.
- H. Y. Huang and C. M. Wang, Phenothiazine: An Effective Molecular Adhesive for Protein Immobilization, J. Phys. Chem. C, 2010, 114, 3560–3567 CAS.
- M. Wainwright, M. N. Byrne and M. A. Gattrell, J. Photochem. Photobiol., B, 2006, 84, 227–230 CrossRef CAS PubMed.
- P. Paul and G. Suresh Kumar, Spectrochim. Acta, Part A, 2013, 107, 303–310 Search PubMed.
- T. J. Peters, Genetics and Medicinal Applications, Academic Press, San Diego, CA, 1996 Search PubMed.
- C. V. Kumar and A. S. Buranaprapuk, Angew. Chem., Int. Ed. Engl., 1997, 36, 2085–2087 CrossRef CAS PubMed.
- S. J. Singer and G. L. Nicolson, Science, 1972, 175, 720–731 CAS.
- W. M. Partridge, Am. J. Physiol., 1987, 252, 157–164 Search PubMed.
- P. Banerjee, S. Pramanik, A. Sarkar and S. C. Bhattacharya, J. Phys. Chem. B, 2009, 113, 11429–11436 CrossRef CAS PubMed.
- X. M. He and D. C. Carter, Nature, 1992, 358, 209–215 CrossRef CAS PubMed.
- T. Peters, Advances in Protein Chemistry, Academic Press, New York, 1985 Search PubMed.
- M. K. Helms, C. E. Peterson, N. V. Bhagavan and D. M. Jameson, FEBS Lett., 1997, 408, 67–70 CrossRef CAS.
- M. El-Kemary, M. Gil and A. Douhal, J. Med. Chem., 2007, 50, 2896–2902 CrossRef CAS PubMed.
- V. Lhiaubet-Vallet, Z. Sarabia, F. Bosca and M. A. Miranda, J. Am. Chem. Soc., 2004, 126, 9538–9539 CrossRef CAS PubMed.
- M. C. Jimenez, M. A. Miranda and I. Vaya, J. Am. Chem. Soc., 2005, 127, 10134–10135 CrossRef CAS PubMed.
- T. Patrice, Photodynamic Therapy, Royal Society of Chemistry, GB, 2004 Search PubMed.
- Y. J. Hu, Y. O. Yang, C. M. Dai, Y. Liu and X. H. Xiao, Biomacromolecules, 2010, 11, 106–112 CrossRef CAS PubMed.
- R. W. Sawbins, Handbook of biological dyes and stains-Synthesis and industrial applications, John Wiley & Sons, New Jersey, 2010 Search PubMed.
- B. K. Hoefelschweiger, A. Duerkop and O. S. Wolfbeis, Anal. Biochem., 2005, 344, 122–129 CrossRef CAS PubMed.
- B. Bhattacharya, S. Nakka, L. Guruprasad and A. Samanta, J. Phys. Chem. B, 2009, 113, 2143–2150 CrossRef CAS PubMed.
- M. R. Eftink and C. A. Ghiron, Biochemistry, 1976, 15, 672–680 CrossRef CAS.
- A. Selva sharma, S. Anandhakumar and M. Ilanchelian, J. Lumin., 2014, 151, 206–218 CrossRef CAS PubMed.
- J. Tian, J. Liu, W. He, Z. Hu, X. Yao and X. Chen, Biomacromolecules, 2004, 5, 1956–1961 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of fluorescence spectroscopy, Springer, New York, 2009 Search PubMed.
- M. Van de Weert and L. Stella, J. Mol. Struct., 2011, 998, 144–150 CrossRef CAS PubMed.
- A. F. M. M. Rahman, S. Bhattacharya, X. Peng and T. Kimura, Chem. Commun., 2008, 1196–1198 RSC.
- J. Liang, Y. Cheng and H. Han, J. Mol. Struct., 2008, 892, 116–120 CrossRef CAS PubMed.
- J. Zhang, Z. Tian, L. Liang, M. Subirade and L. Chen, J. Phys. Chem. B, 2013, 117, 14018–14028 CrossRef CAS PubMed.
- H. A. Benesi and J. H. Hildebrand, J. Am. Chem. Soc., 1949, 71, 2703–2707 CrossRef CAS.
- K. Shanmugaraj, S. Anandakumar and M. Ilanchelian, J. Photochem. Photobiol., B, 2014, 131, 43–52 CrossRef CAS PubMed.
- K. A. Connors, Thermodynamics of pharmaceutical systems: An Introduction for Students of Pharmacy, Wiley-Inter science, 2002 Search PubMed.
- U. Anand, C. Jash and S. Mukherjee, J. Phys. Chem. B, 2010, 114, 15839–15845 CrossRef CAS PubMed.
- E. Lissi, C. Calderon and A. Campos, Photochem. Photobiol., 2013, 89, 1413–1416 CrossRef CAS PubMed.
- Y. J. Hu, Y. Liu and X. H. Xiao, Biomacromolecules, 2009, 10, 517–521 CrossRef CAS PubMed.
- F. F. Tian, F. L. Jiang, X. L. Han, C. Xiang, Y. S. Ge, J. H. Li, Y. Zhang, R. Li, X. L. Ding and Y. Liu, J. Phys. Chem. B, 2010, 114, 14842–14853 CrossRef CAS PubMed.
- G. Zhang, N. Zhao and L. Wang, J. Lumin., 2011, 131, 2716–2724 CrossRef CAS PubMed.
- M. Ilanchelian, C. Retna Raj and R. Ramaraj, J. Inclusion Phenom. Macrocyclic Chem., 2000, 36, 9–20 CrossRef CAS.
- L. Shang, Y. Wang, J. Jiang and S. Dong, Langmuir, 2007, 23, 2714–2721 CrossRef CAS PubMed.
- D. Sarkar, RSC Adv., 2013, 3, 24389–24399 RSC.
- X. Zhao, R. Liu, Z. Chi, Y. Teng and P. Qin, J. Phys. Chem. B, 2010, 114, 5625–5631 CrossRef CAS PubMed.
- J. B. F. Lloyd, Nature, Phys. Sci., 1971, 231, 64–65 CrossRef CAS.
- J. B. F. Lloyd, J. Forensic Sci. Soc., 1971, 11, 83–94 CrossRef CAS.
- J. N. Miller, Recent advances in molecular luminescence analysis, Proceedings of the Analytical Division of the Chemical Society. 1979, vol. 16, pp. 203–208 Search PubMed.
- T. Yuan, A. M. Weljie and H. J. Vogel, Biochemistry, 1998, 37, 3187–3195 CrossRef CAS PubMed.
- S. Naveenraj and S. Anandan, J. Photochem. Photobiol., C, 2013, 14, 53–71 CrossRef CAS PubMed.
- F. Ding, J. Huang, J. Lin, Z. Li, F. Liu, Z. Jiang and Y. Sun, Dyes Pigm., 2009, 82, 65–70 CrossRef CAS PubMed.
- S. M. Kelly, T. J. Jess and N. C. Price, Biochim. Biophys. Acta, 2005, 1751(2), 119–139 CrossRef CAS PubMed.
- B. K. Paul, D. Ray and N. Guchhait, Phys. Chem. Chem. Phys., 2013, 15, 1275–1287 RSC.
- M. Sarkar, S. Shankar Paul and K. K. Mukherjea, J. Lumin., 2013, 142, 220–230 CrossRef CAS PubMed.
- K. M. Naik and S. T. Nandibewoor, J. Lumin., 2013, 143, 484–491 CrossRef CAS PubMed.
- B. K. Paul and N. Guchhait, J. Phys. Chem. B, 2011, 115, 10322–10334 CrossRef CAS PubMed.
- H. Liu, W. Bao, H. Ding, J. Jang and G. Zou, J. Phys. Chem. B, 2010, 114, 12938–12947 CrossRef CAS PubMed.
- S. Neelam, M. Gokara, B. Sudhamalla, D. G. Amooru and R. Subramanyam, J. Phys. Chem. B, 2010, 114, 3005–3012 CrossRef CAS PubMed.
- Y. Zhang, Y. Li, L. Dong, J. Li, W. He, X. Chen and Z. Hu, J. Mol. Struct., 2008, 875, 1–8 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2014 |
Click here to see how this site uses Cookies. View our privacy policy here. 
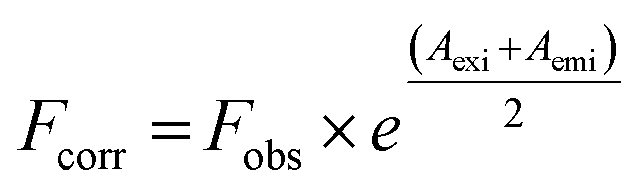
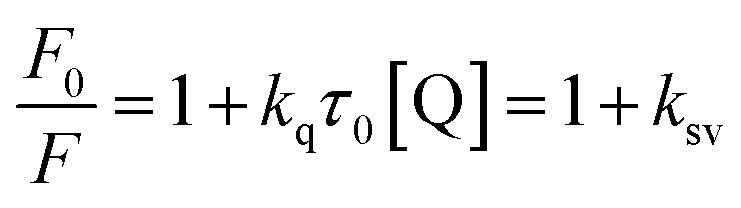
 ] and BSA–AZA [■] complexes.
] and BSA–AZA [■] complexes.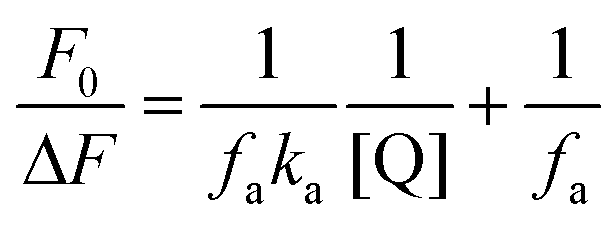
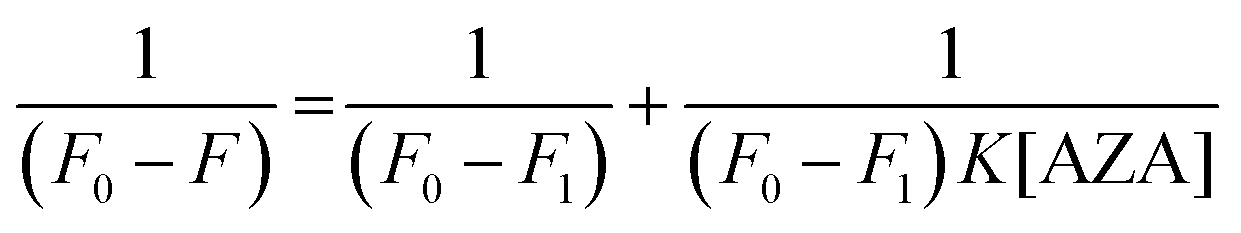
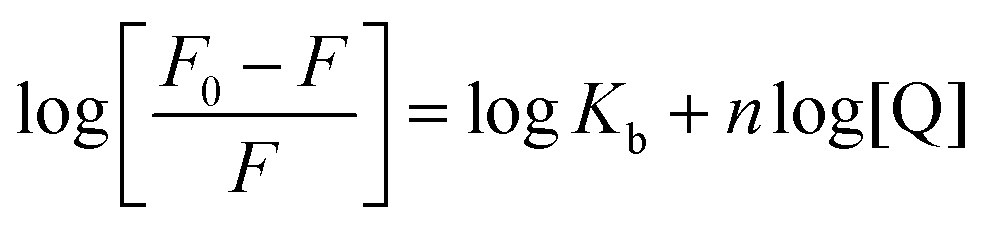
 ] and BSA–AZA complexes [■].
] and BSA–AZA complexes [■].