
Synthesis of monolithic zirconia aerogel via a nitric acid assisted epoxide addition method
Liang Zhonga,
Xiaohong Chen*a,
Huaihe Songa,
Kang Guoa and
Zijun Hu*b
aState Key Laboratory of Chemical Resource Engineering, Beijing Key Laboratory of Electrochemical Process and Technology for Materials, Beijing University of Chemical Technology, Beijing, 100029, P. R. China. E-mail: chenxh@mail.buct.edu.cn; Fax: +86-010-64434916; Tel: +86-010-64434916
bNational Key Laboratory of Advanced Functional Composite Materials, Aerospace Research Institute of Materials & Processing Technology, Beijing 100076, P. R. China. E-mail: huzijun@hotmail.com; Fax: +86-010-68755517; Tel: +86-010-68755517
First published on 15th July 2014
Abstract
Monolithic zirconia aerogels were prepared using zirconium oxychloride (ZrOCl2) as the precursor in the presence of the epoxide (PO) in both ethanol and mixed ethanol–water solutions, respectively. The effects of nitric acid and water contents on the gel formation behavior and the properties of the resulting aerogel were investigated. The results show that at a molar ratio of HNO3/ZrOCl2 of 1.0 and H2O/ZrOCl2 of 10, the translucent monolithic ZrO2 aerogels were obtained and they exhibited a well-developed mesoporous structure and a high specific surface area of 454 m2 g−1. The as-prepared aerogels were composed of mainly amorphous phase and a spot of ZrO2 crystal. After calcination at 750 °C for 2 h, the ZrO2 transformed into tetragonal phase. These results suggested that this HNO3-assisted epoxide addition method was a simple and controllable way to get translucent monolithic zirconia aerogels.
1. Introduction
Mesoporous zirconia (ZrO2) has attracted a lot of attention in recent years because of its unique structures and wide potential applications in catalysts,1–3 catalyst supports4,5 and solid oxide fuel cells (SOFCs).6 Thus, extensive effort has been made to synthesize mesoporous zirconia. According to the types of precursors, it is convenient here to divide these preparation approaches for ZrO2 into two broad groups: alkoxide7–9 and non-alkoxide routes. In alkoxide process, not only are the alkoxides difficult to prepare and handle, but also the sol–gel process of alkoxides are hard to control. Therefore, researchers explored lots of methods using non-alkoxides as precursor, such as alcohol-thermal method,10 alcohol–water-thermal method,11 and the electrolysis method.12 The non-alkoxides used as precursors are cheaper and more manageable than alkoxides. However, their resultant products are usually in forms of powders or highly cracked monoliths. In 2001, Gash13 et al. reported a new sol–gel route to synthesize several transition and main-group metal oxide aerogels using epoxides as proton scavenger from inorganic salts in polar protic solvents. The mechanism of proton-scavenging was reported to occur in two steps: (1) protonation of the epoxide oxygen by the hydrolysis of hydrated metal species and (2) nucleophilic attack of the protonated ring by the nucleophilic anionic.14 As a result, the epoxides undergoes irreversible ring opening. The metal oxide gels prepared with bases like OH− and NH3 are precipitation due to the rapid reaction rate of the base with hydrated metal species.15,16 However, the epoxide is not a strong enough base to induce the formation of precipitation immediately. With time goes, the epoxides consume smoothly the counterion and the proton hydrate in the metal salt solvent. This allows the uniform formation of oligomers, which link together to be sol particles and then lager clusters that subsequently cross-link to form a homogeneous gel. Christopher et al.6,17 synthesized yttria-stabilized zirconia aerogel using the “epoxide addition” method. Using the epoxide (PO) as the gel initiator is more convenient to get a higher specific area aerogel than using the oxetane. However, the gelation occurred rapidly. It only took several minutes in pure water system while just a few seconds in alcohol–water system. The gel time is too short to provide a chance for preparing aerogel composite materials via this approach. In addition, under high temperature supercritical drying the as-prepared aerogel is coarse and dense. Therefore, it is a great challenge to control the process of gel–sol and integrate the mesoporous structures into a ZrO2 monolith using epoxide addition method. Much less work has been devoted to synthesize the monolithic ZrO2 aerogel using this process.Herein, we present a controllable strategy for the preparation of translucent and monolithic zirconia aerogel without any stabilizer via the epoxide addition method by adjusting the water and acid contents. The process of gel–sol can be easily controlled by the molar ratio of HNO3/ZrOCl2 and H2O/ZrOCl2. The gel time can be largely prolonged, which helps to form a more interconnected network and also provides more chance of preparing aerogel composites. Additionally, the as-prepared zirconia aerogel displayed a well-developed mesoporous structure and a high specific surface area of 454 m2 g−1. Besides, the metastable tetragonal ZrO2 can be obtained after calcinated at 750 °C for 2 h.
2. Experimental sections
2.1 Synthesis of ZrO2 Aerogel
ZrO2-precursor gels were prepared by the addition epoxide method followed by supercritical drying in ethanol. Zirconium oxychloride octahydrate (ZrOCl2·8H2O), propylene oxide (PO), and HNO3 (65% w/w) were used as received. Two beakers were prepared: one with ZrOCl2·8H2O, ethanol, H2O and HNO3; the other with ethanol and PO. The total molar ratio of ZrOCl2, H2O, HNO3 and PO was kept constant at 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:1:6. The two solutions were mixed and vigorously stirred until the vortex created by the stirring disappeared owing to gelation. Furthermore, a water bath (20 °C) was needed in the process of hybrid. After 24 h aging, the alcogels were washed with ethanol every 24 h and renewed 2–3 times in order to make sure the solvents in the gel network were fully replaced by ethanol. The resultant alcogels were then supercritically dried by ethanol in an autoclave. The autoclave was pressurized to 2.5 MPa with purity nitrogen. Then, the temperature was heated to 538 K, while the pressure was controlled at 7.2 MPa. After keeping this supercritical state for 1 h, the fluid was slowly released to atmospheric pressure. When the autoclave was cooled to room temperature, the resulting zirconia aerogel was obtained. To determine the role of the acid in gel formation for this system, zirconia aerogels were prepared from varying acid content using a similar way described above. The aerogels with varied acid contents are noted as Nx and X (=0, 0.6, 1.0, 1.2) is the molar ratio of acid to zirconium. Similarly, the aerogels with varied water contents are called Hx and X (=2, 10, 20) is the molar ratio of water to zirconium. The resulting aerogels were calcined at 350 °C, 450 °C, 550 °C, 750 °C, 850 °C, and 1000 °C in air for 2 h with a heating rate 5.0 °C·min−1, respectively.
:10:1:6. The two solutions were mixed and vigorously stirred until the vortex created by the stirring disappeared owing to gelation. Furthermore, a water bath (20 °C) was needed in the process of hybrid. After 24 h aging, the alcogels were washed with ethanol every 24 h and renewed 2–3 times in order to make sure the solvents in the gel network were fully replaced by ethanol. The resultant alcogels were then supercritically dried by ethanol in an autoclave. The autoclave was pressurized to 2.5 MPa with purity nitrogen. Then, the temperature was heated to 538 K, while the pressure was controlled at 7.2 MPa. After keeping this supercritical state for 1 h, the fluid was slowly released to atmospheric pressure. When the autoclave was cooled to room temperature, the resulting zirconia aerogel was obtained. To determine the role of the acid in gel formation for this system, zirconia aerogels were prepared from varying acid content using a similar way described above. The aerogels with varied acid contents are noted as Nx and X (=0, 0.6, 1.0, 1.2) is the molar ratio of acid to zirconium. Similarly, the aerogels with varied water contents are called Hx and X (=2, 10, 20) is the molar ratio of water to zirconium. The resulting aerogels were calcined at 350 °C, 450 °C, 550 °C, 750 °C, 850 °C, and 1000 °C in air for 2 h with a heating rate 5.0 °C·min−1, respectively.
2.2 Physical Characterization
The morphologies and structures of the samples were investigated via transmission electron microscope (TEM, Hitachi H-800) and high-resolution transmission electron microscope (HRTEM, JEOL JEM-2100). Nitrogen sorption isotherms were measured with ASAP 2020 (Micromeritics, USA), and the sample was degassed at 150 °C overnight. The specific surface area and pore size distribution were calculated by the Brunauer–Emmett–Teller (BET) method and Barrett–Joyner–Halenda (BJH) method, respectively. The products were characterized by X-ray powder diffraction (XRD) on a Rigaku D/max-2500B2+/PCX system operating at 40 kV and 20 mA with Cu Kα radiation (λ = 1.5406 A°, 2θ = 10–90°). Raman spectroscopy was carried out using a 785 nm laser (Aramis, Jobin Yvon).3. Results and discussion
3.1 Effects of reaction parameters on the formation of ZrO2 gels
Addition of epoxide (PO) to ethanol or aqueous ethanol solutions of ZrOCl2 resulted in the formation of monolithic gels that varied from translucent to opaque depending on the reaction conditions. Assisted by the proton-scavenging properties of epoxide, the hydrated metal species are induced to undergo hydrolysis and condensation reactions to from small oligomers. Then, these oligomers grow to be sol particles and lager clusters in sequence. Finally, the gels with interconnected network are produced. Table 1 shows the effects of the molar ratio of acid to zirconium (N) and water to zirconium (H) on the gel time and the characteristic of ZrO2 gels. The gel time (tgel) is the interval between addition of the epoxide and the point at which the sols no longer flow when the reaction containers are tilted.| Sample | Nb | Hc | tgel (min) | Morphology |
|---|---|---|---|---|
| a The molar ratio of epoxide to zirconium.b The molar ratio of acid to zirconium.c The molar ratio of H2O to zirconium. | ||||
| H2 | 1 | 0 | 16 | Opaque white |
| H20 | 1 | 20 | 40 | Translucent white |
| N0 | 0 | 10 | ∼5s | Opaque white |
| N0.6 | 0.6 | 10 | 2.5 | Opaque white |
| N1.0 (H10) | 1.0 | 10 | 25 | Translucent white |
| N1.2 | 1.2 | 10 | 45 | Translucent white |
With the N value increasing from 0 to 1.2, the tgel raise form 5 s to 45 min as well as the ZrO2 gels vary from opaque to translucent. Basically, the formation of gels lies on the hydrolysis and successive condensation of oligomers. In the acidic condition, the rate of the sequential condensation can be effectively influenced by the acid concentration, because these reactions are positively charged and stabilized by the repulsive electrostatic interaction in a fashion similar to that observed for colloidal particles-dispersed system.18,19 Therefore, with the use of higher acid concentration, the rate of condensation can be obviously slowed down. What is more, the relative hydrolysis and condensation rates play an important role on the different wet-chemical characteristics of the sol–gel products.20 When N is 0.6, fast relative rates of hydrolysis and condensation generally lead to white-opaque appearance, indicating the presence of localized condensation. If the N value increased, i.e., for N = 1, fast hydrolysis with low condensation yield translucent polymeric alcogels with a well cross-linked network.21–23 The obtained gels are translucent and bouncy. However, when N = 1.2, the resulting gels are easy to shrink and crack, illustrating that excessive consumption of epoxy by nitric acid results in that clusters do not grow large enough to connect to become an extended network. Moreover, at much higher value of N, gelation will be completely suppressed. These gelation behaviors are similar with the result of Sul et al.24 who prepared the alkoxide-drived gels under the different acid concentration.
Likewise, the effects of the molar ratio of water to zirconium (H) on the gel time and the appearance of gels are also investigated by varying the value of H from 2 to 20. To avoid the effect of acid on the gelation behavior, the value of N is kept constant at 1.0 in this process. As shown in Table 1, the tgel raises from 16 to 40 min with the value of H increasing from 2 to 20. The transparency of gel is also varied from opaque to translucent with the increase of the value of H. The increase of gel time may be a result of the reduced nucleophilicity of the counterion (Cl−) in water versus ethanol, leading to a slower condensation reaction. Baumann et al.25 also attributed this reasoning to similar changes for the epoxide-initiated gelation of AlCl3·6H2O in ethanol versus water. Besides the nucleophilicity of the counterion, the dielectric constants of reaction media should also be considered. It has been recognized that the dielectric constants of water and ethanol are 80.4 and 25.0 at 20 °C, respectively.26 The dielectric constants of mixed water–ethanol solution are higher with more water. A higher dielectric constant corresponds to a higher solubility, which predicts less clusters numbers and lower growing rates of clusters. This is similar with the “nucleation” and “growth” of crystal in crystallography. It seems to provide another reasonable explanation for why the gel time of H2 is shorter than that of H20. Gash et al. 13used this hypothesis to explain the effect of different solvent on the gel time, when they prepared Cr2O3 aerogel by epoxide addition method. Above all, the gel time and the appearance of gels can be controlled by acid concentration and solvent employed.
3.2 Morphologies and structures of ZrO2 aerogels
Fig. 1 shows the photographs of typical zirconia aerogels. Although the obtained aerogel N0.6 is opaque monolith, it is easy to grind to fine powders. The aerogel N1.0 is translucent and relatively rigid monolith, which is not easily crushed by hand. The aerogel N1.2 is cracked and broke apart, indicating the networks of gel are destroyed during the aerogel drying process. Beside all that, aerogel H2 are cracked opaque monoliths. | ||
| Fig. 1 The photographs of zirconia aerogels: (a) N0.6, (b) N1.0, (c) N1.2 and (d) H2. | ||
The TEM images of aerogel N0.6, N1.0 and H2 are shown in Fig. 2a–c. The degree of network and size of the cluster varied depending on the contents of nitric acid and water. The opaque aerogel N0.6 prepared with fast reaction conditions (Fig. 2a) shows less network and larger clusters than the translucent aerogel N1.0 prepared with relatively slow reaction conditions (Fig. 2b). This microscopy results support the proposal, set earlier, that nitric acid suppresses the presence of localized condensation resulting in small clusters and fine skeleton formation, which also can been confirmed by SEM image shown in Fig. 2f. Aerogel H10 has similar cluster sizes with N0.6. This shows that high nucleophilicity of the counterion and low solubility of sol particles in pure ethanol result in the greater clusters growth. Fig. 2d and e provides a fine representation of the size, shape, connectivity of the clusters. It appears that these particles are relative spheres with a diameter in the 5–10 nm range. They seem to connect to each other to form the lager clusters, in further, to form the network of aerogels. Moreover, it is revealed crystallization has occurred in the skeleton structure of aerogel N1.0. The crystallization size was measured to be about 5 nm.
 | ||
| Fig. 2 TEM images of zirconia aerogels: (a) N0.6, (b) N1.0, (c) H2, (d) HRTEM of N1.0 and (e) enlarged HRTEM image of the selected area marked by a box in (d); (f) the SEM image of N1.0. | ||
3.3 Surface Area and pore structure of ZrO2 aerogels
Fig. 3a shows nitrogen adsorption/desorption isotherms of the aerogels N0, N0.6, N1.0, and N1.2, respectively. All samples exhibit hysteresis at relatively high P/Po, which can be classified as Type IV according to the IUPAC nomenclature. The presence of hysterisis loop, caused by the capillary condensation in the mesopores (2–50 nm), suggests the resulting aerogels are mesoporous materials.27 | ||
| Fig. 3 Nitrogen adsorption–desorption isotherms for ZrO2 aerogels: (a) N0, N0.6, N1.0, and N1.2. The insert in (a) shows the pore size distributions of N0, N0.6, N1.0, and N1.2; (b) H2, H10 and H20.The insert in (b) shows the pore size distributions of H2, H10 and H20. | ||
The BJH pore size distributions of N0, N0.6, N1.0, and N1.2 are present in the insert of Fig. 3a. The most probable pore size is nearly identical for the N0 and N0.6. However, the most probable pore size is found to shift towards larger pore size with higher acid concentration. The aerogels N0.6, N1.0 and N1.2 have pore size distribution centered at 14.8 nm, 18.3 nm and 31.6 nm, respectively. With higher acid concentration, highly branched network is easier to be formed by clusters. Presumably the more branched the gel network is, the larger interconnected clusters may form larger pore. The surface area, pore volume and mesopore fraction for the zirconia aerogels are summary in Table 2. The aerogel N1.0 exhibits the maximum specific surface area (454 m2 g−1) and pore volume (1.8 ml g−1) among all the as-prepared aerogels. The large specific surface area and large pore volume are attributed to the absence of localized condensation, resulting in more branched networks. This is supported by the TEM data, discussed earlier. Furthermore, the mesopore fraction of N1.0 has the maximum value of 97.1%, and that of N0.6 is 66.2%, which suggests more mesopores are formed with higher acid concentration. However, continuing to increase N to 1.2, the specific surface area and pore volume are 406 cm2 g−1 and 1.5 ml g−1, respectively. In addition, the mesopore fraction of N1.2 decreases to 54.7%. These decreases may result from the formation of a weakly cross-linked structure by clusters, which is easy to collapse and agglomerate.
| Sample | SA BET (m2 g−1) | Pore volume (ml g−1) | Mesopore fractiona (%) |
|---|---|---|---|
| a The ratio of the pore volume of pores mesoporous (2–50 nm) to totally pore volume of pores between 1.7 nm and 300.0 nm width determined by BJH method. | |||
| N0 | 113 | 0.5 | 84.1 |
| N0.6 | 226 | 0.6 | 66.2 |
| N1.0 (H10) | 454 | 1.8 | 97.1 |
| N1.2 | 407 | 1.5 | 54.7 |
| H2 | 185 | 0.5 | 66.7 |
| H20 | 406 | 2.5 | 59.5 |
The water content was also an important factor affecting the pore structures. As shown in Fig. 3b, the as-prepared aerogels also display a Type IV isotherm with a closed desorption branch hysteresis loop, which is the typical characteristic of mesopore. The trend of most probable pore size for varying acid concentration is similar with that for varying water content. The translucent aerogel H10 have a considerably higher surface area than opaque aerogel H2, which suggests water, improving the solubility of reaction media and reducing the nucleophilicity of the counterion, slowing down the growth of clusters. Thus, the pore structures of as-prepared aerogels are tailored via addressing the nitric acid and water contents.
3.4 Effects of heat treatment on the structures of ZrO2 aerogel
As well known, calcination can result in the growth and transformation of zirconia crystallite. To investigate the effect of temperature on the crystallographic evolutions for as-prepared zirconia, XRD patterns of zirconia N1.0 calcinated at 350–1000 °C in air for 2 h with a heating rate 10 °C min−1, respectively, and the results are given in Fig. 4a. | ||
| Fig. 4 XRD patterns of aerogel N1.0 calcinated at different temperature: (a) calcinated at 350–1000 °C, respectively; (b) aerogel N1.0 calcinated at 750 °C. The XRD pattern for standard cubic and tetragonal ZrO2 are shown as lines and points with different colors in (b); (c) aerogel N1.0 calcinated at 850 °C. The XRD pattern for standard monoclinic ZrO2 is also shown as lines and points. | ||
It can be seen that aerogel N1.0 without calcination has broad diffraction pattern which is characteristic of amorphous zirconia. However, there is a weak diffraction peak at approximately 50°, indicating that a spot of crystallization has occurred during supercritical drying.21 This is consistent with the HRTEM results discussed earlier. With the increase of the calcination temperature, the broad diffraction peak become sharpened gradually, reflecting an evidently crystallite growth of the zirconia samples. Moreover, several new weak diffraction peaks are also observed, and these peaks are sharpened constantly with the increase of calcination temperature. It can be seen in Fig. 4b that the major peaks are at 30°, 35°, 50°, and 60° with weak peaks at 62°, 74°, and 82° for the zirconia sample calcined at 750 °C. These diffraction peaks are very close to the values of tetragonal (t) phase (PDF #50-1089). Thus, it can be deduced that the t-phase zirconia is obtained. However, as shown in Fig. 4b, the XRD pattern for standard cubic (c) and tetragonal (t) phase ZrO2 are similar and no splitting is observed at the aforementioned 2θ. So the assignment of cubic and tetragonal phases based solely on the XRD is very difficult. It has been well documented that Raman spectroscopy, sensitive to polarizability of the oxygen ions, is recognized as a powerful tool for identifying different polymorphs of metal oxides. According to group theory, monoclinic (m-ZrO2), tetragonal (t-ZrO2) and cubic (c-ZrO2) phase of zirconia are expected to have 18(9Ag + 9Bg), 6(1A1g + 2B1g + 3Eg) and 1T2g Raman active modes, respectively.28–32 Therefore, Raman spectroscopy can easily distinguish between cubic phase and tetragonal. As shown in Fig. 5, the Raman spectrum have bands at 181, 265, 317, 470, and 641 cm−1 for zirconia N1.0 calcinated at 750 °C, which are assigned to the Raman-active modes for the tetragonal phase of ZrO2.28,30,33 However, the single Raman peak at 490 cm−1 for cubic zirconia (ref. 33) does not appear. Thus, the metastable t-phase below the m-t transition temperature (approximately 1150 °C) is obtained when the as-prepared zirconia aerogel is heated. Fig. 4c shows the XRD pattern of as-prepared products at 850 °C. This figure indicates that a mixture of monoclinic (m) and tetragonal (t) of zirconia is obtained. The Raman spectrum (Fig. 5) has bands at 177, 189, 220, 305, 331, 345, 378, 475, 497, 534, 561, 615, and 634 cm−1 for zirconia N1.0 calcinated at 850 °C, which are approximately the same as reported by the previous works.34–36 They can be identified to Raman active modes of m-ZrO2. In addition, the band at 256 cm−1 can also be observed for zirconia N1.0 calcinated at 850 °C, illuminating the presence of t-ZrO2. This agrees with the XRD result. When calcination temperature was increased to 1000 °C (Fig. 4a), the amount of m-phase is considerably increased. In addition, as observed the selected area marked by a box in Fig. 4a, the intensity of peak corresponding to t-phase increases proportionally with that of m-phase, increasing the calcined temperature from 850 °C to 1000 °C, which indicates that the m-phase and t-phase ZrO2 may be formed simultaneously at a relatively high temperature.
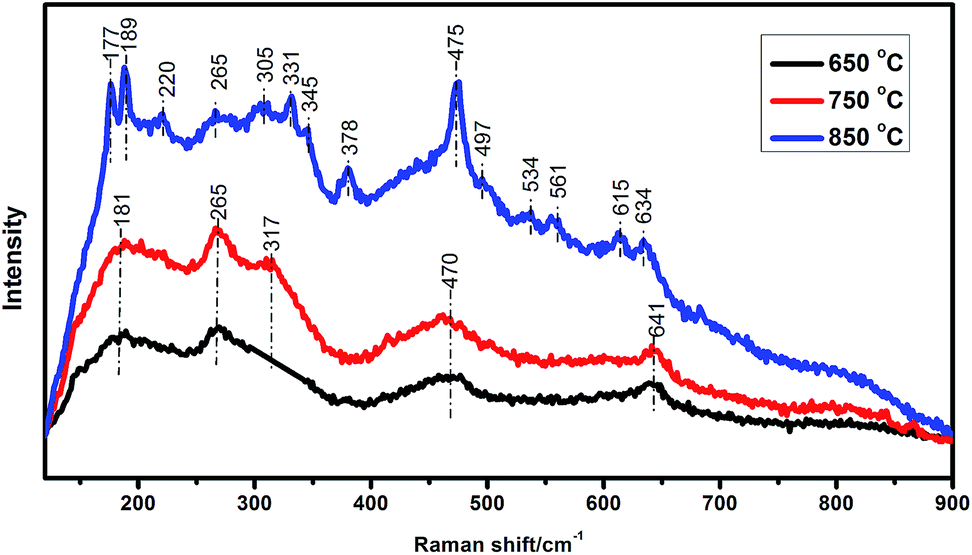 | ||
| Fig. 5 Raman spectra of aerogel N1.0 calcined at 650 °C, 750 °C, and 850 °C, respectively. | ||
4. Conclusions
Zirconia monolithic aerogels had been successfully prepared via the epoxide addition method by adjusting the solvent and acid content. The opaque gels are always obtained at a lower molar ratio of acid to zirconium and water to zirconium, while with the increase of the acid and water contents, the obtained gels are translucent. When the HNO3/ZrOCl2 molar ratio is 1.0, the gel time is prolonged to 25 min, which is not only helpful for forming a stable gel with an extended network but also allows for preparation of aerogel composite materials. However, overmuch acid and water contents decrease the strength of gels structure leading to the occurrence of cracks and shrinkage during supercritical drying process. Additionally, the acid and water contents were found to be critical factors in the morphology and microporous structure of the resulting aerogels. At a molar ratio of HNO3/ZrOCl2 of 1.0 and H2O/ZrOCl2 of 10, the resultant ZrO2 aerogel monolith exhibited a well-developed mesoporous structure and a high specific surface area of 454.04 m2 g−1. However, when more acids were used (e.g. the molar ratio of HNO3/ZrOCl2 was 1.2), the as-prepared aerogels were cracked and broke apart as well as their specific surface area decreased. This is attributed to excessive consumption of epoxy by nitric acid decreases the cross-linking, resulting in the network of gels are damaged during the aerogel drying process.Heat treatment at different temperatures for 2 h plays an important role in the crystallographic evolutions for the as-prepared zirconia. The resulting aerogels were mainly amorphous phase and contained a spot of crystal. With the increase of the calcination temperature, an evidently tetragonal phase growth of the zirconia had occurred. The combined XRD and Raman results suggested that tetragonal ZrO2 were obtained calcinated at 750 °C for 2 h. However, the as-prepared zirconia calcinated at 850 °C or higher temperature were a mixture of monoclinic (m) and tetragonal (t).
Acknowledgements
This work was supported by the National Natural Science Foundation of China (51202009 and 51272016), New Teachers' Fund for Doctor Stations, Ministry of Education of China (20120010120004), and Foundation of Excellent Doctoral Dissertation of Beijing City (YB20121001001).Notes and references
- Y. Y. Huang, B. Y. Zhao and Y. C. Xie, Appl. Catal., A, 1998, 173, 27–35 CrossRef CAS.
- I. Mejri, M. Younes, A. Ghorbel, P. Eloy and E. Gaigneaux, Stud. Surf. Sci. Catal., 2006, 162, 953–960 CrossRef CAS.
- G. Pajonk and A. El Tanany, React. Kinet. Catal. Lett., 1992, 47, 167–175 CrossRef CAS.
- L. Chen, J. Hu and R. M. Richards, ChemPhysChem, 2008, 9, 1069–1078 CrossRef CAS PubMed.
- Y. Sun and P. Sermon, Top. Catal., 1994, 1, 145–151 CrossRef CAS.
- C. N. Chervin, B. J. Clapsaddle, H. W. Chiu, A. E. Gash, J. H. Satcher Jr and S. M. Kauzlarich, Chem. Mater., 2006, 18, 4865–4874 CrossRef CAS.
- A. Bedilo and K. Klabunde, Nanostruct. Mater., 1997, 8, 119–135 CrossRef CAS.
- C. Stöcker, M. Schneider and A. Baiker, J. Porous Mater., 1995, 2, 171–183 CrossRef.
- C. Stöcker, M. Schneider and A. Baiker, J. Porous Mater., 1996, 2, 325–330 CrossRef.
- J. Hu, Y. Cao and J. Deng, Chem. Lett., 2001, 30, 398–399 CrossRef.
- Z. G. Wu, Y. X. Zhao, L. P. Xu and D. S. Liu, J. Non-Cryst. Solids, 2003, 330, 274–277 CrossRef CAS PubMed.
- Z. Zhao, D. Chen and X. Jiao, J. Phys. Chem. C, 2007, 111, 18738–18743 CAS.
- A. E. Gash, T. M. Tillotson, J. H. Satcher Jr, L. W. Hrubesh and R. L. Simpson, J. Non-Cryst. Solids, 2001, 285, 22–28 CrossRef CAS.
- A. E. Gash, T. M. Tillotson, J. H. Satcher Jr, J. F. Poco, L. W. Hrubesh and R. L. Simpson, Chem. Mater., 2001, 13, 999–1007 CrossRef CAS.
- Y. Y. Huang, B. Y. Zhao and Y. C. Xie, Appl. Catal., A, 1998, 172, 327–331 CrossRef CAS.
- V. Quaschning, J. Deutsch, P. Druska, H. J. Niclas and E. Kemnitz, J. Catal., 1998, 177, 164–174 CrossRef CAS.
- C. N. Chervin, B. J. Clapsaddle, H. W. Chiu, A. E. Gash, J. H. Satcher Jr and S. M. Kauzlarich, Chem. Mater., 2005, 17, 3345–3351 CrossRef CAS.
- B. Derjaguin, Acta Physicochim. URSS, 1941, 14, 633–662 Search PubMed.
- E. J. W. Verwey, J. Phys. Chem., 1947, 51, 631–636 CrossRef CAS.
- J. Livage, M. Henry and C. Sanchez, Prog. Solid State Chem., 1988, 18, 259–341 CrossRef CAS.
- D. A. Ward and E. I. Ko, Chem. Mater., 1993, 5, 956–969 CrossRef CAS.
- B. E. Yoldas, J. Mater. Sci., 1986, 21, 1080–1086 CrossRef CAS.
- C. Stöcker and A. Baiker, J. Non-Cryst. Solids, 1998, 223, 165–178 CrossRef.
- D. J. Suh and T. J. Park, Chem. Mater., 1996, 8, 509–513 CrossRef CAS.
- T. F. Baumann, A. E. Gash, S. C. Chinn, A. M. Sawvel, R. S. Maxwell and J. H. Satcher Jr, Chem. Mater., 2005, 17, 395–401 CrossRef CAS.
- G. Akerlof, J. Am. Chem. Soc., 1932, 54, 4125–4139 CrossRef CAS.
- K. S. W. Sing, D. H. Everett, R. A. W. Haul, L. Moscou, R. A. Pierotti and J. Rouquerol, Pure Appl. Chem., 1985, 57, 603–619 CrossRef CAS.
- V. G. Keramidas and W. B. White, J. Am. Ceram. Soc., 1974, 57, 22–24 CrossRef CAS PubMed.
- W. B. White, Mater. Res. Bull., 1967, 2, 381–394 CrossRef CAS.
- P. Bouvier, H. Gupta and G. Lucazeau, J. Phys. Chem. Solids, 2001, 62, 873–879 CrossRef CAS.
- G. Teufer, Acta Crystallogr., 1962, 15, 1187–1187 CrossRef CAS.
- G. A. Kourouklis and E. Liarokapis, J. Am. Ceram. Soc., 1991, 74, 520–523 CrossRef CAS PubMed.
- C. Phillippi and K. Mazdiyasni, J. Am. Ceram. Soc., 1971, 54, 254–258 CrossRef CAS PubMed.
- Y. Jing-Lin, J. Guo-Chang, Y. Song-Hua, M. Jin-Chang and X. Kuang-Di, Chin. Phys. Lett., 2001, 18, 991 CrossRef.
- K. Kanade, J. Baeg, S. Apte, T. Prakash and B. Kale, Mater. Res. Bull., 2008, 43, 723–729 CrossRef CAS PubMed.
- D. A. Zyuzin, S. V. Cherepanova, E. M. Moroz, E. B. Burgina, V. A. Sadykov, V. G. Kostrovskii and V. A. Matyshak, J. Solid State Chem., 2006, 179, 2965–2971 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2014 |