
Structural electrochemistry. Chronopotentiometric responses from rising compacted polypyrrole electrodes: experiments and model
Abstract
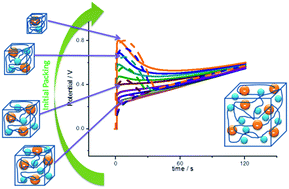
Reduced self-supported polypyrrole films have been oxidized by flow of a constant anodic current (galvanostatic experiments) in aqueous solution. The chronopotentiometric responses obtained from films reduced at high cathodic potentials present an initial peak attributed to the relaxation of packed conformational structures. This is a structural electrochemical response. The influence on this peak of the reduction potential, the electrolyte concentration or the anodic current was studied. The conformational relaxation model gives a good theoretical description of the experimental chronopotentiometric responses under the influence of the studied variables. The experimental evolution of the consumed electrical energy and the separation of the oxidation–relaxation and oxidation–diffusion components are also described by the theoretical model. The energy consumed during the oxidation reaction is a sensor of both the electrolyte concentration and the driving current, also described by the model.
- This article is part of the themed collection: Nanoscience and nanotechnology in electrochemistry
 Please wait while we load your content...
Please wait while we load your content...

