
“The green side of the moon: ecofriendly aspects of organoselenium chemistry”
Stefano Santoro
a,
Juliano B. Azeredo
b,
Vanessa Nascimento
b,
Luca Sancineto
c,
Antonio L. Braga
b and
Claudio Santi
*c
aDepartment of Organic Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91, Stockholm, Sweden
bLaboratório de Síntese de Substâncias de Selênio Bioativas, Centro de Ciências Físicas e Matemáticas, Departamento de Química, Universidade Federal de Santa Catarina, 88040-900, Florianópolis, SC, Brazil. E-mail: braga.antonio@ufsc.br
cDipartimento di Scienze Farmaceutiche, Università di Perugia, Via del Liceo 1, 06134, Perugia, Italy. E-mail: claudio.santi@unipg.it
First published on 14th July 2014
Abstract
Organoselenium chemistry has proven to be a powerful tool for organic synthesis over several decades. Nevertheless, the use of selenating reagents has often been limited by a generally bad reputation surrounding selenium toxicity and its potential impact on the environment. In this review we would like to stress some aspects that will encourage the reader to discover an unexpected “green side” to this element and the chemistry connected with its organic derivatives.
 Stefano Santoro | Stefano Santoro was born in Campobasso, Italy, in 1980. He received his Ph.D. degree at the University of Perugia in 2009 (with Prof. Claudio Santi and Prof. Marcello Tiecco). Between 2006 and 2007 he has been visiting Ph.D. student at Aarhus University, in the group of Prof. Karl Anker Jørgensen. In 2010 he moved to Stockholm University as a postdoctoral researcher with Prof. Fahmi Himo. His main research interests are in the areas of asymmetric catalysis, C–H functionalizations and mechanistic investigations by means of DFT calculations. |
 Juliano B. Azeredo | Juliano Braun de Azeredo was born in Santa Maria, Brazil, in 1987. He received his B.Sc. in Chemistry in 2009 from the Federal University of Santa Maria (South Brazil). Immediately he moved to Federal University of Santa Catarina (Brazil) where he completed his M.Sc. degree. Currently, he is completing his Ph.D. degree at the same university under supervision of Prof. Antonio Luiz Braga. Between 2013–2014, during the period of five months, he received a fellowship to join Professor Claudio Santi's group. His main interests are focus on the development of green methodologies for the synthesis of organochalcogen compounds. |
 Vanessa Nascimento | Vanessa Nascimento was born in Marau (RS State), Brazil. In 2009 she obtained her B.Sc. degree in Chemistry from Universidade Federal de Santa Maria. Then, she moved to Universidade Federal de Santa Catarina, where received her M. Sc degree in 2011. She is currently developing her Ph. D. activities under the supervision of Professor Antonio L. Braga at the same university. In 2013–2014 she was awarded with a fellowship to join Professor Claudio Santi's group over a period of five months. Her research mainly focus in the synthesis of new organoselenium compounds with relevant biological activities. |
 Luca Sancineto | Luca Sancineto PhD, graduated in 2008 with a bachelor's degree in Pharmacy at the university of Perugia with a comprehensive final grade of summa cum laude. In 2012 he obtained his PhD degree in medicinal chemistry under the supervision of Prof. Oriana Tabarrini and Prof. Violetta Cecchetti. Presently, he his PostDoc at the Organic Chemistry section of the Department of Pharmaceutical Sciences of the university of Perugia under the supervision of Prof. Claudio Santi, supported by a fellowship of the “Consorzio Interuniversitario Nazionale Metodologie e Processi Innovatici di Sintesi, C.I.N.M.P.I.S.”. His research interest lies in the development of selenium-containing novel antiviral agents targeting HIV. |
 Antonio L. Braga | Antonio Luiz Braga obtained his BSc in 1982 from the Universidade Federal de São Carlos. He received his MSc and PhD degrees from the Universidade de São Paulo in 1984 and 1989, working under the direction of Prof. Comasseto. In 1985 he obtained a position at the Universidade Federal de Santa Maria-RS as Assistant Professor. During 1990 to 2009 he occupied a position at the same University as Associate Professor. In 2009, Prof. Braga moved to Florianópolis where he became a faculty member of the Universidade Federal de Santa Catarina-SC, as Professor of Organic Chemistry, position that he still holds at the present date. |
 Claudio Santi | Claudio Santi is professor of Organic Chemistry at Perugia University. He obtained his PhD in 1996 under the supervision of professor Marcello Tiecco and after a postdoctoral stay with Professor Thomas Wirth at Basel University (Switzerland), he becomes assistant professor in Perugia and then aggregated professor (2005) associate professor (2013) obtaining the habilitation as full professor in 2014. He was invited as visiting professor to the University of Kracov (PL), the School of Chemistry of Cardiff (UK) and the Jan Dlugosz University in Csestokowa (PL). His main research interests are in the areas of chalcogen chemistry. |
1. Introduction
Although selenium was discovered in the 18th century by the Swedish chemist Jöns Jacob Berzelius, its widespread application in organic chemistry began in the 1970s, when the selenoxide elimination reaction was reported as a convenient method for the synthesis of olefins.1 The possibility to promote a large number of different reactions, including C–C and carbon-heteroatoms bond formations and cyclofunctionalization, makes organoselenium compounds very interesting reagents in modern synthesis. These reaction, moreover, are often characterized by high levels of chemo-, regio- or stereoselectivity. Organoselenium compounds can act, as nucleophiles, electrophiles or radicals, in a wide variety of direct functional groups transformations, potentially avoiding protection–deprotection steps that are particularly detrimental in the design of a green chemical procedure.2In addition, the discovery of a series of selenoproteins as an important class of mammalian antioxidant enzymes increased significantly the interest in the chemistry of this element. Some organoselenium compounds have been recently investigated also for their biological activities, such as antioxidant, anticancer, anti-inflammatory, and antiviral, due to their capability to act as mimetic of selenium-containing enzymes.3 It is, therefore, increasingly common to find in the literature examples of organoselenium compounds as final products, rather than just as intermediates in multi-step synthesis.
On the other hand, since the definition of the concept of green chemistry in 1990s,4 the development of chemical procedures aiming at reducing the use and the generation of hazardous substances developed dramatically. In recent years, environmental issues have been highlighted in national and international media and a more sustainable development is on the agenda of most of the government leaders.5 The “green chemistry approach”, aiming at achieving sustainability at the molecular level, is nowadays considered with great interest by chemical and pharmaceutical industries, as well as an affirmed and well-developed field of study and research in the academia.
In 2009, some of us highlighted that many of the criteria contained in the 12 rules of green chemistry can be addressed by selenium-based catalysis.6
This idea involves also the biological properties of selenoproteins, the natural catalysts selected by the evolution for the regulation of several redox balances in living systems, and could be considered a seminal starting point for the development of a branch of recent research that will be covered by this review article.7
The key aspects for the development of a greener approach to the synthesis and the use of the organoselenium compounds can be collected in three main areas: the use of alternative solvents and/or reaction media (such as ionic liquids, glycerol and water), the use of non-conventional and energy-saving sources of activation (microwaves, ultrasounds and electrochemistry) and organoselenium derivatives as catalysts with particular regard to the bio-inspired applications.
In this review we intend to stress the potential use of organoselenium reagents for the development of greener reactions, taking inspiration from some examples that already present seminal characteristic connected and/or related to some of the twelve “green rules”. It is not our intention to comprehensively review all the treated topics and for this reason we chose selected examples, mainly focusing on our own research interests. Moreover, since some reactions presented here could fit in more than one section of this review, we had to take arbitrary decisions in these cases.
We also included a brief paragraph, at the end of this review, discussing the toxicity of selenium and selenium-containing compounds. In our view, this could stimulate a more realistic approach on the definition of the chemistry involving these derivatives as final products, intermediates, reagents and wastes.
2. Organoselenium reagents in non-conventional media
2.1 Reactions in water
Water is a low-cost, safe, non-toxic and often reusable reaction media which can positively contribute to prevent the production of non-recyclable organic wastes. Several organic reactions have been demonstrated to proceed in good yields and selectivity also in aqueous media,8 and recently a number of new organoselenium reagents were designed to perform selenium mediated reactions in water or in “on water” conditions.9Among them, an interesting class of selenates was prepared through a 100% atom economical oxidative insertion of elemental zinc into commercially available PhSe-halides,10,11a,b and subsequently used in several efficient nucleophilic reactions.11 These nucleophilic reagents are not water soluble and have been used in “on water” conditions for different synthetic purposes: nucleophilic substitutions on sp2 and sp3 carbons,11a,b,e ring opening reactions on epoxides,11a,b,12 as well as Michael-type additions to unsaturated ketones (Scheme 1).11d In all cases the reaction effected in water suspension showed higher rates compared to the same reaction effected in THF, indicating an activation mechanism similar to those described by Sharpless in the theory of the “on water” conditions.12
 | ||
| Scheme 1 Reactivity of PhSeZnX (X = Cl or Br). | ||
More recently, some of us reported the synthesis of selenol esters by reaction of acyl chlorides 1 with PhSeZn-halides in “on water” conditions. Surprisingly, the nucleophilic substitution of the acyl chloride by nucleophilic selenium was in every case faster than the hydrolysis, affording aryl and alkyl phenylselenol esters 2 in moderate to excellent yields (ranging from 20% to 97%, Scheme 2).13 The chemoselectivity was proposed to be associated to a concerted mechanism in which the zinc ion acts as a Lewis acid coordinating the carbonyl oxygen and directing the nucleophilic selenium attack. The recyclability of the aqueous medium was also investigated and it was demonstrated that it is possible to obtain good yields for more than ten consecutive runs, by simply neutralizing the pH after each run.
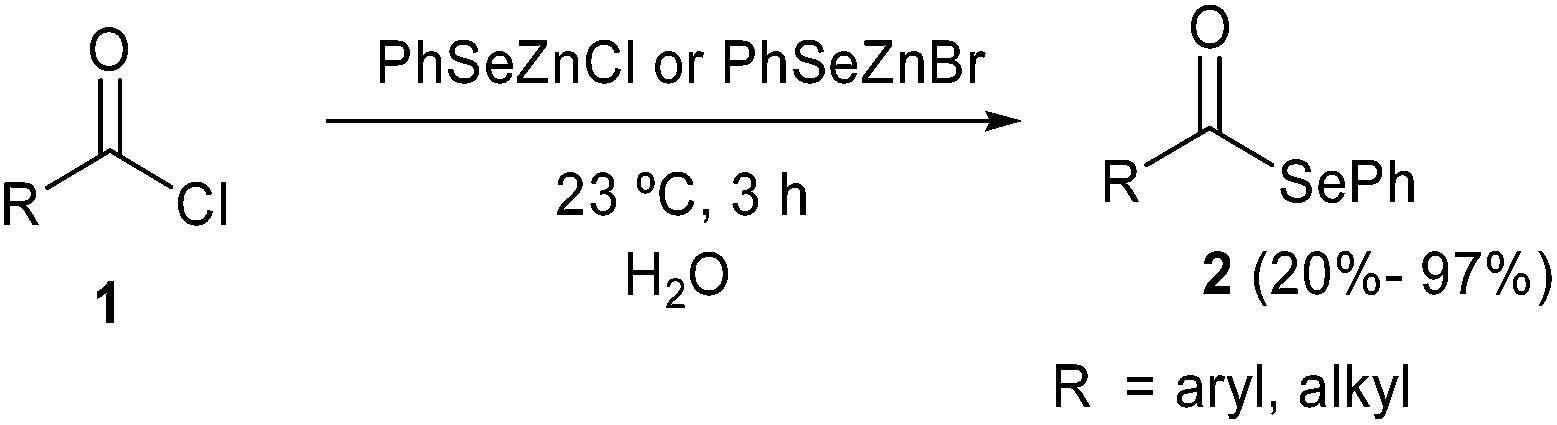 | ||
| Scheme 2 Synthesis of selenol esters using PhSeZn-halides. | ||
Ranu and co-workers reported the synthesis of aryl selenides 4 and vinyl selenides 5 using aryl iodides or vinyl bromides, diphenyl diselenide 6, zinc dust and catalytic amounts of copper nanoparticles in aqueous medium, as shown in Scheme 3.14 A variety of aryl selenides were prepared, bearing electron-donating as well as electron-withdrawing groups on the aromatic ring of the aryl halides. For the synthesis of vinyl selenides, it was observed that (E)-vinyl bromides provide (E)-vinyl selenides in a stereoselective way, while (Z)-vinyl bromides afford mixtures of (E) and (Z) geometric isomers. It is worth mentioning that the copper nanoparticles can be recovered and reused for subsequent reaction runs. A different mechanism is probably involved in the reaction of vinyl halides and PhSeZnCl in “on water” conditions. The substitution at the vinylic carbon occurred with retention of the configuration of the starting material with the only exception of olefins conjugated with a carbonyl group. In the latter case the reaction resulted to be stereoconvergent affording the (Z)-isomer starting from either (E)- or (Z)-halides. DFT calculations demonstrated the effect of the zinc coordination on this selectivity.11e
 | ||
| Scheme 3 Copper catalyzed preparation of aryl- and vinyl selenides. | ||
Recently the development and application of novel strategies for the synthesis of homogeneous proteins largely involved selenium intermediates and their reaction in aqueous media. As an example, the non-natural amino acid phenylselenocysteine (PhSec) has been efficiently used for the chemoselective introduction of N-methyl, N-acetyl or phosphoric moieties into proteins.
The reaction is promoted in very mild conditions by hydrogen peroxide (40 minutes at room temperature in pH 8.5 Tris buffer) and converts PhSec to dehydroalanine. This latter is then allowed to react with lysine derivatives or with phosphorylating agents affording the final posttranslational modification mimics through a Michael addition reaction (Scheme 4).15
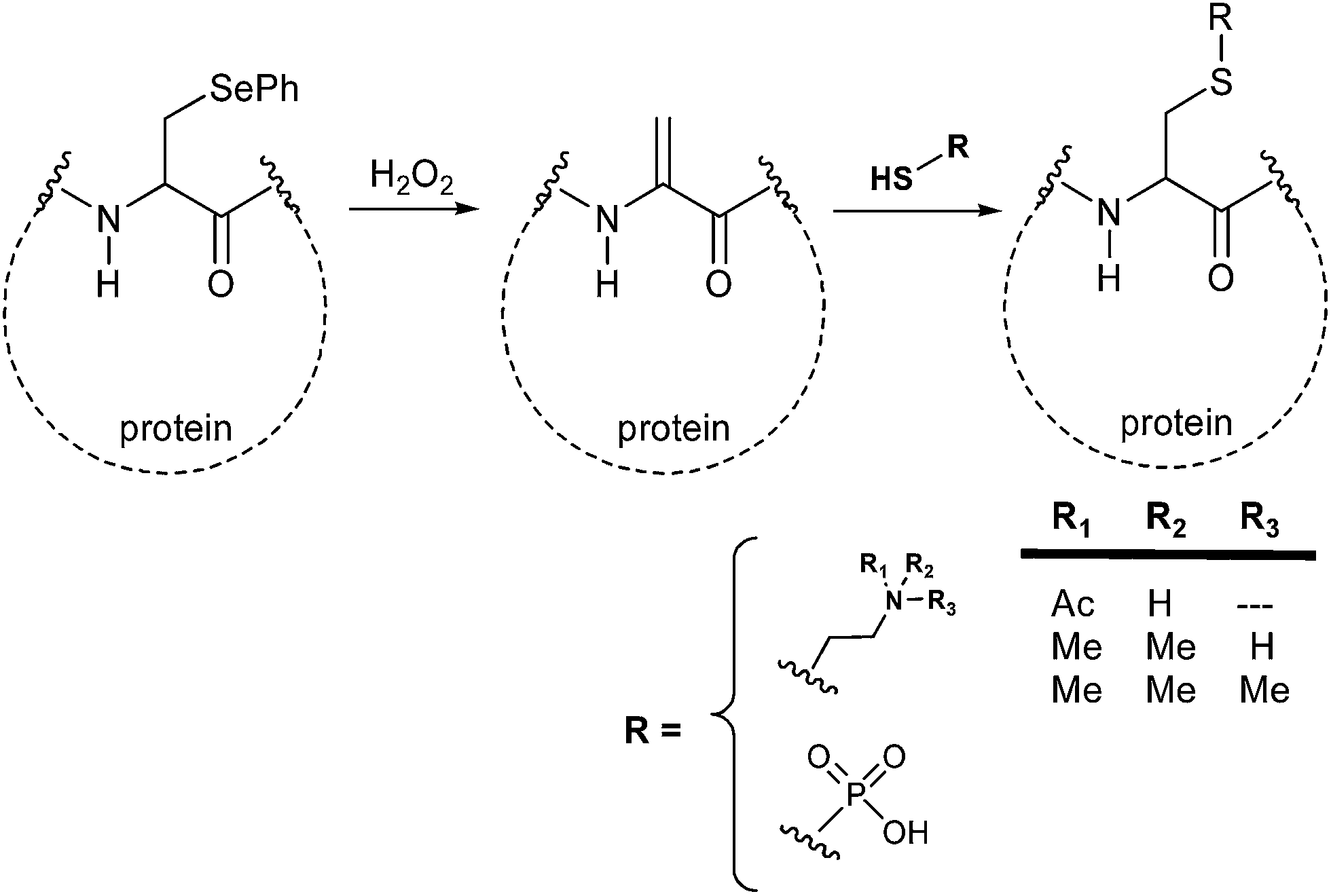 | ||
| Scheme 4 Phenylselenocysteine in the synthesis of posttranslational modification mimics. | ||
Danishefsky and co-workers reported the synthesis of a proline diselenide as a ligation agent in the functionalization of peptides containing a sterically demanding amino acid on the C-terminal. The reaction proceeds in high yields in a 7.4–7.6 pH buffer. Also, the deselenation occurred in aqueous media in the presence of dithiotreitol (DTT) at pH 5.0–6.0 (Scheme 5).16
 | ||
| Scheme 5 Seleno-proline mediated ligation. | ||
The introduction of an allyl selenide moiety as a suitable metathesis substrate enables a number of protein surface modifications difficult to achieve through other approaches. The installation of extended Se-allylselenocysteine (Seac) on protein surface is obtained by reacting Seac-acrylamide in the presence of DTT and a catalytic amount of DMF in a buffered aqueous medium (Scheme 6).17 Similarly, protected β-selenol phenylalanine derivatives can be incorporated into peptides and used in ligation reactions with peptide thioesters. From the resulting peptide the selenium moiety can be removed using tris(2-carboxyethyl)phosphine (TCEP) and DTT in a one-pot procedure to afford an Ala residue.18
 | ||
| Scheme 6 Protein surface modification by reaction with Seac-acrylamide. | ||
In a different way a similar functionalization can be effected following a biomimetic approach via a conjugated addition of allyl selenoate to dehydroalanine.19
Chalcogen-assisted cross-metathesis can be performed in aqueous medium. Allyl selenide-containing proteins showed a remarkably higher reactivity than the corresponding sulfur derivatives. The difference in reactivity may be attributed to the softness of selenium, which makes the coordination to ruthenium even more favorable than the sulfur coordination. As an example, the reaction between functionalized protein and allyl alcohol required very mild conditions, affording almost quantitative conversion in only 15 minutes at room temperature. Efficient cross metathesis was demonstrated for the reaction of unhindered allyl selenide-containing proteins with several substrates including carbohydrates, oligo(ethylene glycols), allyl acetamides and alkenes with electron-withdrawing ammonium salts.
The application of organoselenium intermediates in water has been recently reported for the preparation of CdSe quantum dots. Luminescent aqueous colloids of mercaptopropionic acid capped cadmium selenide nanoparticles can be prepared using SeO2 as selenium source. The oxide reacts with water to afford seleninic acid and this is reduced by thiol to the corresponding sulfur selenide (RSSeSR) that, due to its instability, acts as source of Se ions suitable for the preparation of CdSe nanoparticles.20
2.2 Reactions in ionic liquids (IL)
Ionic liquids can be employed as alternative solvents in organic synthesis due to some favorable properties such as non-volatility, non-flammability, thermal and chemical stability and recyclability. Essentially for the same reasons they are considered environmentally benign reaction medium.21 In recent years, many advances have been obtained in the application of IL in organic synthesis, and several examples of their use as a reaction medium in the preparation of selenium compounds have been reported.22A wide range of selenol esters 2 were prepared from the reaction between diorganoaryl diselenides and acyl chlorides 1, using [bmim]PF6 as the best renewable medium after the screening of several different ionic liquids (Scheme 7).
 | ||
| Scheme 7 Preparation of selenol esters in ionic liquid (IL). | ||
The reaction can be activated by CuO nanopowder23 or Zn dust,24 but in this latter case the corresponding products 2 was obtained generally in higher yields (73–98%) and shorter reaction times at room temperature. The evaluation of the ionic liquids recyclability demonstrated that in both cases the solvent could be reused for four consecutive runs without appreciable loss of effectiveness.
Similarly, the synthesis of diaryl selenides 6 using Zn powder can be conveniently effected in [bmim]BF4 under neutral reaction conditions, as shown in Scheme 8. The ionic liquid, which can be reused for 5 runs, showed higher performance compared to other conventional solvents.25 These compounds were also prepared in good yields by a cross-coupling reaction between aryl and alkyl bromides and diselenides, by using a catalytic amount of CuO nanopowder and 2 equivalents of KOH in the same IL that showed also the same recyclability.26
 | ||
| Scheme 8 Preparation of diorganoyl selenides. | ||
The reaction of electrophilic selenium species and boron derivatives was described by Lenardão and co-workers as an alternative protocol for the preparation of diaryl selenides 8 in [bmim]PF6 or [bmim]BF4 (Scheme 9).27 The selective synthesis of these compounds were realized in a catalyst-free process, using aryl boronic acids 9 or potassium aryltrifluoroborate 10 through a substitution reaction with arylselenenyl halides (chlorides or bromides) 7. Yields were generally good, ranging from 73% to 96%.
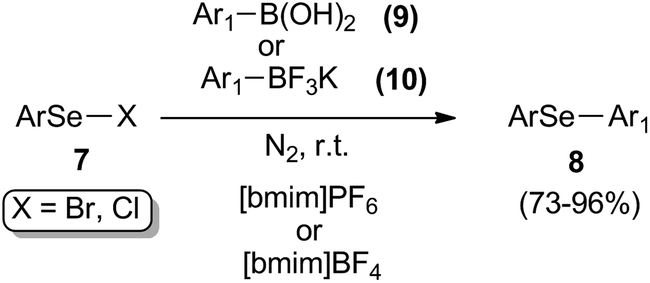 | ||
| Scheme 9 Preparation of diaryl selenides. | ||
2.3 Reactions in glycerol and polyethylene glycol (PEG)
In the last few years, relevant efforts have been devoted to the development of non-conventional fuels, essentially due to the increase of oil price and to the need to replace traditional fuels for environmental reasons.28 In this context, the production and use of biodiesel are steadily growing. The major side product of biodiesel production is glycerol, whose production largely exceeds the market demand.29 Due to its large availability and to its physical and chemical properties, glycerol is generally considered as an environmental friendly solvent.30 The use of glycerol as a solvent has also been reported in the field of organoselenium chemistry, especially in metal catalyzed coupling reactions.31For instance, glycerol was employed in a very efficient copper-catalyzed cross-coupling reaction between aryl boronic acids and diaryl diselenides, in the presence of a stoichiometric amount of DMSO as oxidant (Scheme 10).25 The best results were obtained with CuI as catalyst and a range of diaryl selenides were prepared in good to excellent yields. Recyclability studies were also performed, showing that the glycerol–CuI system could be recovered and reused for successive cycles with limited loss in efficiency.
 | ||
| Scheme 10 Synthesis of diaryl selenides employing glycerol as solvent. | ||
Alternatively, polyethylene glycol (PEG) can be used as an environmentally benign reaction medium in organic chemistry, due to its easy recyclability. The use of this solvent in the synthesis of organoselenium compounds has also been described. As an example, the selective α-selenylation of aldehydes and ketones was reported to occur in the presence of a solid-supported catalyst (KF/Al2O3) and PEG-400 as recyclable medium.32 By this protocol, several 2-phenylseleno aldehydes and ketones could be synthesized in good to excellent yields. This protocol was also applied to the synthesis of the bactericide agent 2-phenylselenocitronellal 11, as shown in Scheme 11.
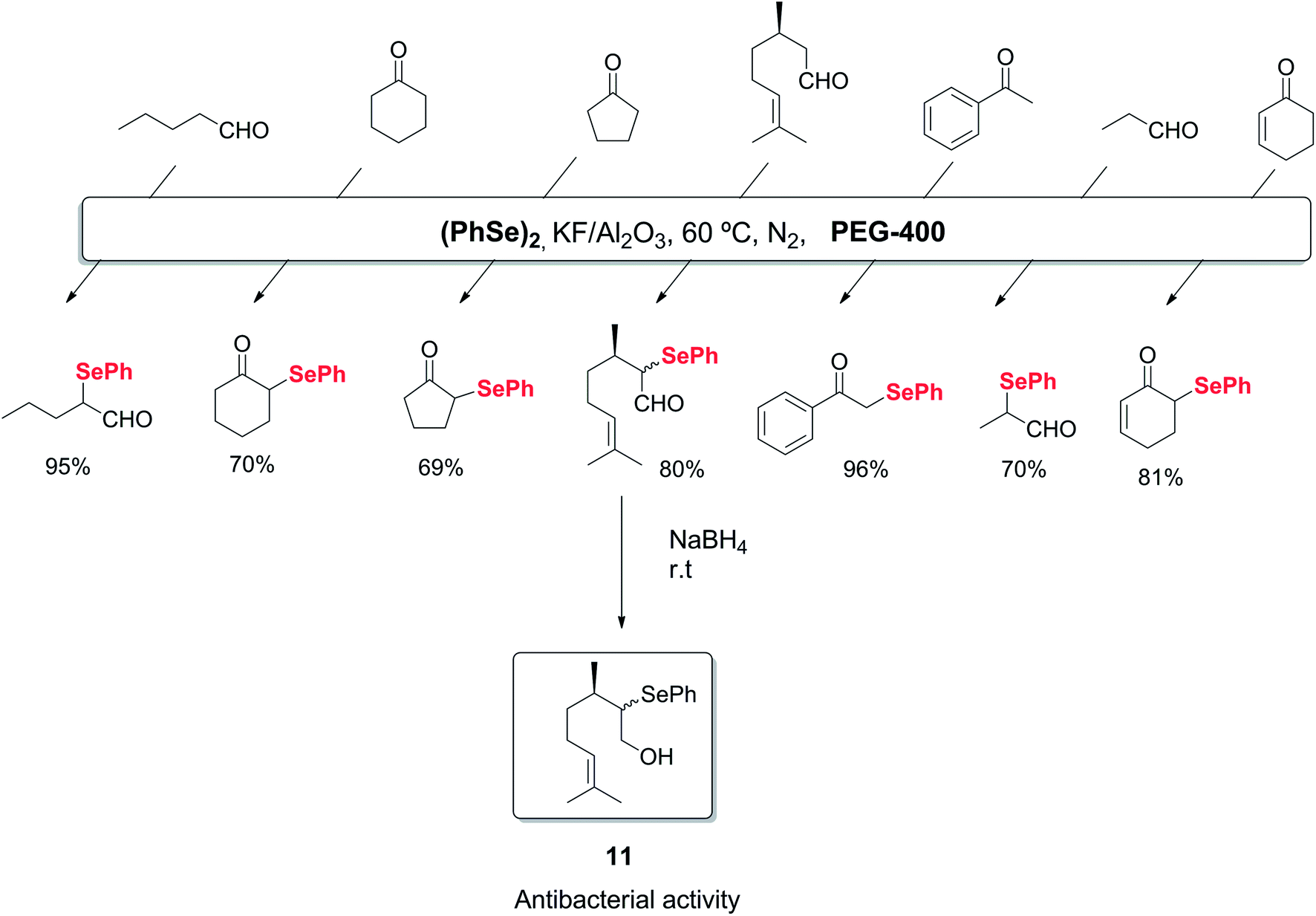 | ||
| Scheme 11 Electrophilic addition of selenium species to aldehydes and ketones. | ||
2.4 Reactions in solvent-free conditions
Solvent-free reaction conditions have been recognized as an important strategy for improving sustainability of organic transformations.33 This approach is continuously developing since it allows the reduction of waste generation while avoiding at the same time some drawbacks related to flammability, toxicity and safety of common organic solvents. In this sense, new protocols for the synthesis of organoselenium compounds avoiding the use of solvents have been recently reported in the literature.34Vinylic bis-selenides 12 could be obtained from the addition of diselenides to alkynes using Pd(PPh3)4 as catalyst, under solvent-free reaction conditions (Scheme 12).35 The advantages of this methodology, beside the usual benefits of not using an organic solvent, are in the quantitative yields, high stereoselectivity of (Z)-isomer formation and shorter reaction times.
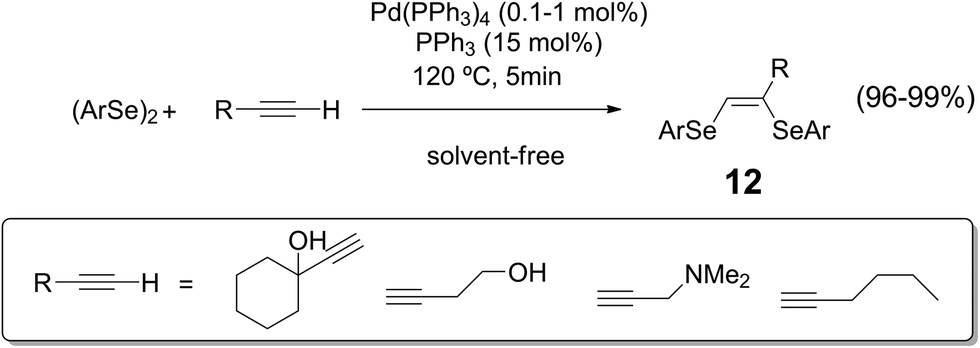 | ||
| Scheme 12 Pd-catalyzed synthesis of vinyl bis-selenides. | ||
A variety of selenol esters 2 were synthesized in solvent-free conditions, by reacting acyl chlorides and diselenides in the presence of zinc (Scheme 13).29 The reactions completed in only 2 minutes under microwave irradiation. In addition, selenocarbonates 13, an interesting organoselenium protecting group, were also prepared using this ecofriendly approach.36
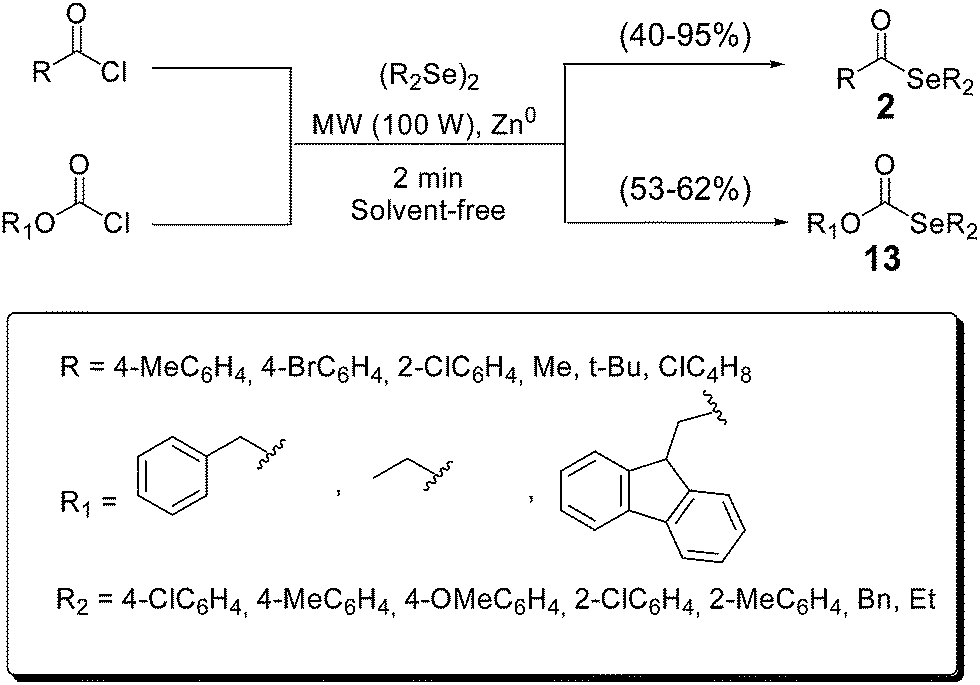 | ||
| Scheme 13 Solvent-free synthesis of selenol esters and selenocarbonates. | ||
Recently, a new solvent-, ligand-, and metal-free protocol for the synthesis of unsymmetrical diaryl chalcogenides 15 was developed (Scheme 14).27 Diaryl dichalcogenides and diazonium tetrafluorborates 14 react in the presence of KOH and neutral alumina in a ball-milling system, affording the corresponding products in short times and good yields.
 | ||
| Scheme 14 Solvent-free synthesis of unsymmetrical selenides. | ||
3. Application of different sources of energy in organoselenium chemistry
3.1 Microwave-assisted reactions
The use of microwave as an alternative source of heating has received much attention in organic chemistry.37 Although it is debated whether microwave heating is actually more energy-efficient than conventional heating or not,38 the use of microwave can still have some potential benefits. These advantages, resulting mainly from the decrease of wall effects, include shorter reaction times, limited side products formation and better reproducibility compared to the relative conventional process.39Microwave irradiation has found wide application in the synthesis of organoselenium compounds.40
A copper-based catalytic system for the preparation of diaryl chalcogenides 18 under microwave heating was reported by Kumar and Engman (Scheme 15). Aryl halides 16 and dichalcogenides 17 could undergo cross-coupling reactions mediated by CuI/bipyridyl as catalyst in the presence of magnesium and DMF as solvent. With this methodology the reaction times could be reduced from 2–3 days to 6–8 hours.41
 | ||
| Scheme 15 Microwave-assisted synthesis of diaryl chalcogenides. | ||
Another valuable approach to obtain unsymmetrical diaryl chalcogenides (S, Se, Te) under microwave irradiation makes use of diaryl dichalcogenides 20, aryl diazonium fluoroborates 21 and dimethyl carbonate (DMC) in the presence of zinc dust (Scheme 16).42 This protocol could provide a wide range of substituted diaryl chalcogenides 18 in high yields and short reaction times.
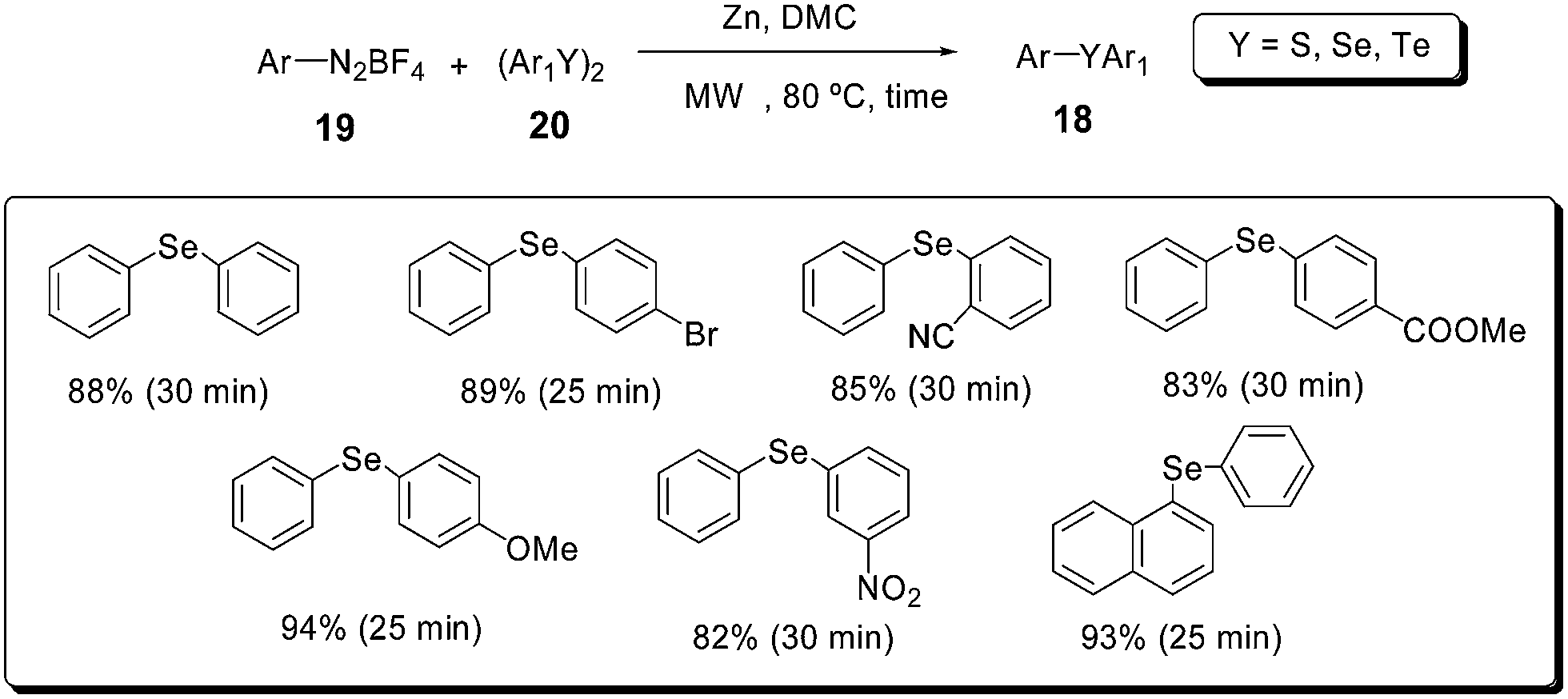 | ||
| Scheme 16 Zinc mediated microwave-accelerated synthesis of diaryl selenides. | ||
In 2012, some of us reported the C–Y bond formation (Y = S, Se, Te), catalyzed by CuO nanoparticles, for the synthesis of symmetrical diorganoyl dichalcogenides 21 (Scheme 17).43 Starting from aryl iodides, elemental chalcogen, KOH and DMSO as solvent, a variety of compounds (21) could be synthesized in good to excellent yields and short reaction times (7–15 minutes).
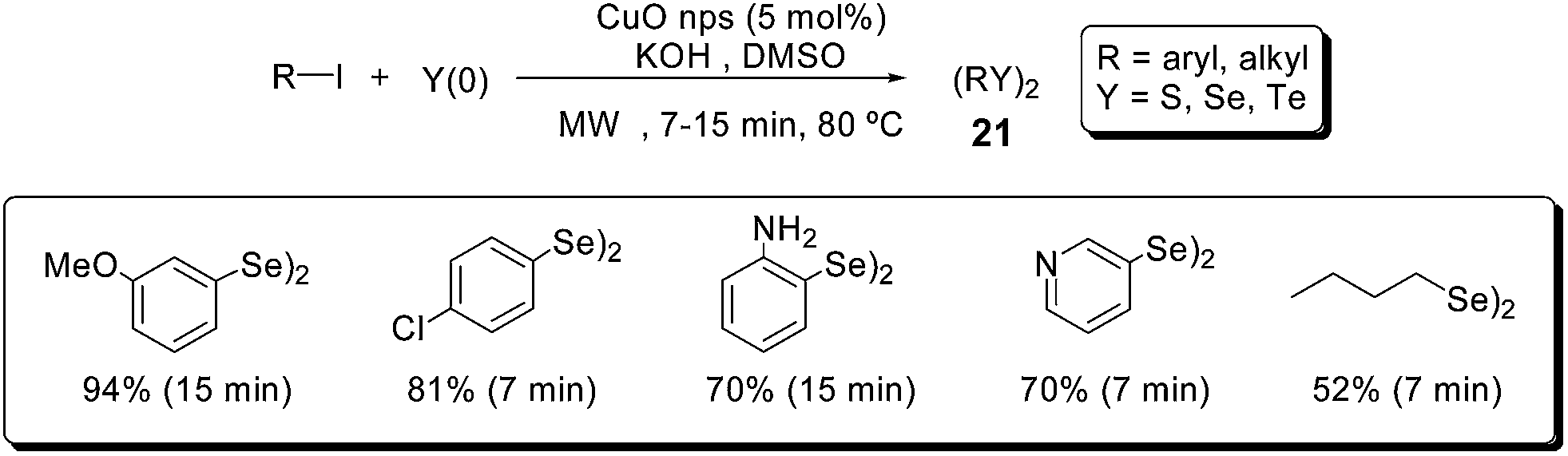 | ||
| Scheme 17 Synthesis of diorganoyl diselenides under microwave irradiation. | ||
Chiral β-seleno amides 22 were applied as ligands for the microwave-accelerated palladium-catalyzed asymmetric allylic alkylation reaction (Scheme 18).44 The reaction of rac-1,3-diphenyl-2-propenyl acetate 23 with different malonates (24), catalyzed by the enantiopure β-seleno amide palladium complexes, provides the respective alkylated products 25 in good yields and enantiomeric excess, in very short reaction times (2–4 minutes).
 | ||
| Scheme 18 Microwave-assisted asymmetric allylic alkylations with selenium-based chiral ligands. | ||
More recently, some of us described a fast microwave-accelerated synthesis of 3-chalcogenyl indoles employing solvent-free conditions. The electrophilic chalcogenyl reagents were synthetized in situ from the corresponding dichalcogenides in the presence of DMSO as stoichiometric oxidant and iodine as catalyst.45
3.2 Sonochemistry
Another environmentally benign source of energy used in organic chemistry is the ultrasound irradiation.46 Ultrasound waves promote the cavitation phenomena, which is the creation of gaseous or vaporous bubbles in an irradiated sample.47 This causes an increase in temperature and pressure inside the cavities, raising the mass transfer of the reaction media. The advantages of the use of this technique are the reduction of reaction times, high selectivity, easy experimental manipulation and the possibility to work at room temperature.A few examples of the application of ultrasounds in organoselenium chemistry have been reported in the literature.48 In 2011, Cheng and co-workers developed an epoxides ring-opening by diphenyl diselenide under ultrasound irradiation (Scheme 19). The reaction was performed in the presence of a base, sodium dithionite and a PEG-400–H2O mixture as solvent. The corresponding β-hydroxyselenides were obtained in high regioselectivities and yields, with reaction times ranging from 40 to 50 minutes. Moreover, the reaction conditions proved to be effective on a large scale.
 | ||
| Scheme 19 Selenium epoxide ring-opening under ultrasound irradiation. | ||
3.3 Electrochemically promoted reactions
Electrochemistry, though little used in organoselenium chemistry, can be considered among the form of non-conventional activation energies. In particular, this approach was used to generate electrophilic reagents starting from diphenyl diselenides.The procedure avoids the use of stoichiometric amount of halogens improving the atom economy of the process and reducing the side reaction due to the presence of a stoichiometric amount of a nucleophilic counter ion.
Konstantinovic and co-workers reported that in the presence of a catalytic amount of an electrolyte (tetraethylammonium bromide or calcium chloride) the PhSe+ species can be generated by anodic oxidation and used to promote cyclofunctionalization reactions starting from alkenols (Scheme 20).49
 | ||
| Scheme 20 Electrochemically promoted cyclofunctionalization. | ||
4. Organoselenium reagents as catalysts
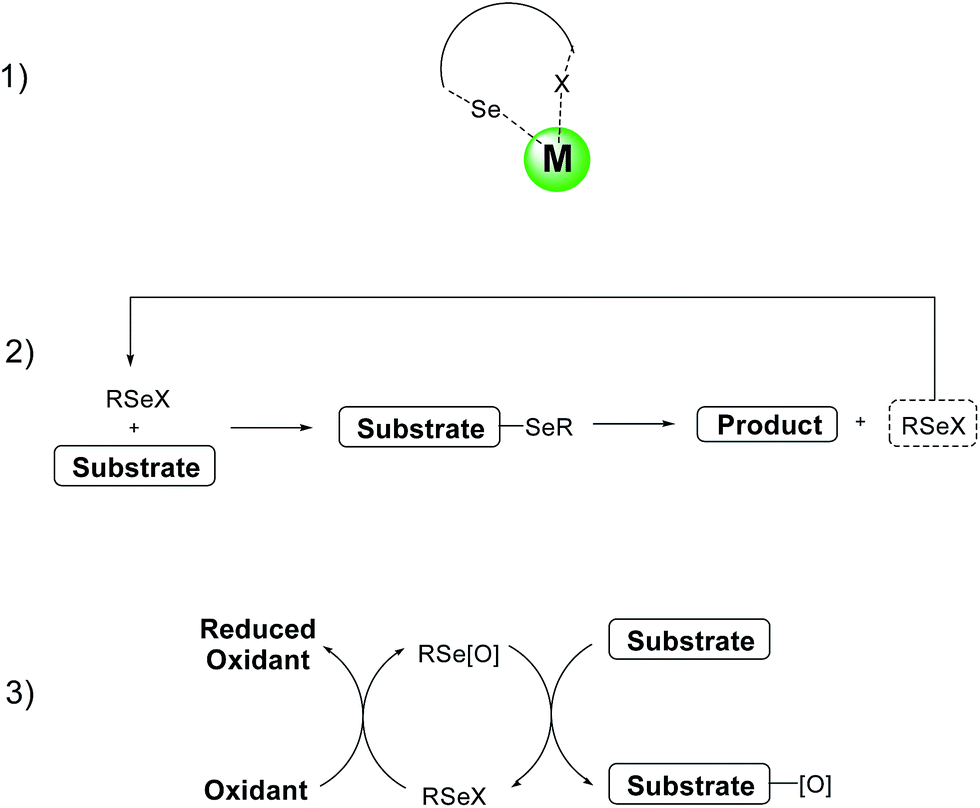
The use of selenium based reagents as catalysts has been recently reviewed in a number of articles6,50 and book chapters.7a For this reason this paragraph will only briefly cover some recent applications, together with some examples selected on the basis of their historic relevance.Organoselenium derivatives can be used as catalysts or co-catalysts in three different ways: (1) as ligands in metal-catalyzed reactions; (2) as organocatalysts for selenenylation–deselenenylation processes; (3) as oxygen carrier in oxidation reactions (Scheme 21).
 | ||
| Scheme 21 Catalytic applications of organoselenium compounds. | ||
4.1 Selenides and diselenides in metal-catalyzed reactions
Even if the use of organoselenium compounds as ligands in catalysis dates back to 1975,51 the first efficient applications only came twenty years later, when Uemura first and several other groups later developed and employed new optically pure selenium based reagents in a number of different asymmetric reactions. Some representative examples are reported in Scheme 22: (a) the rhodium-catalyzed hydrosilylation for the enantioselective reduction of ketones or olefins,52 (b) the enantioselective formation of new C–C bond by the diethyl zinc addition to aldehydes,53 and (c) the palladium-catalyzed allylic alkylation (Tsuji–Trost reaction).54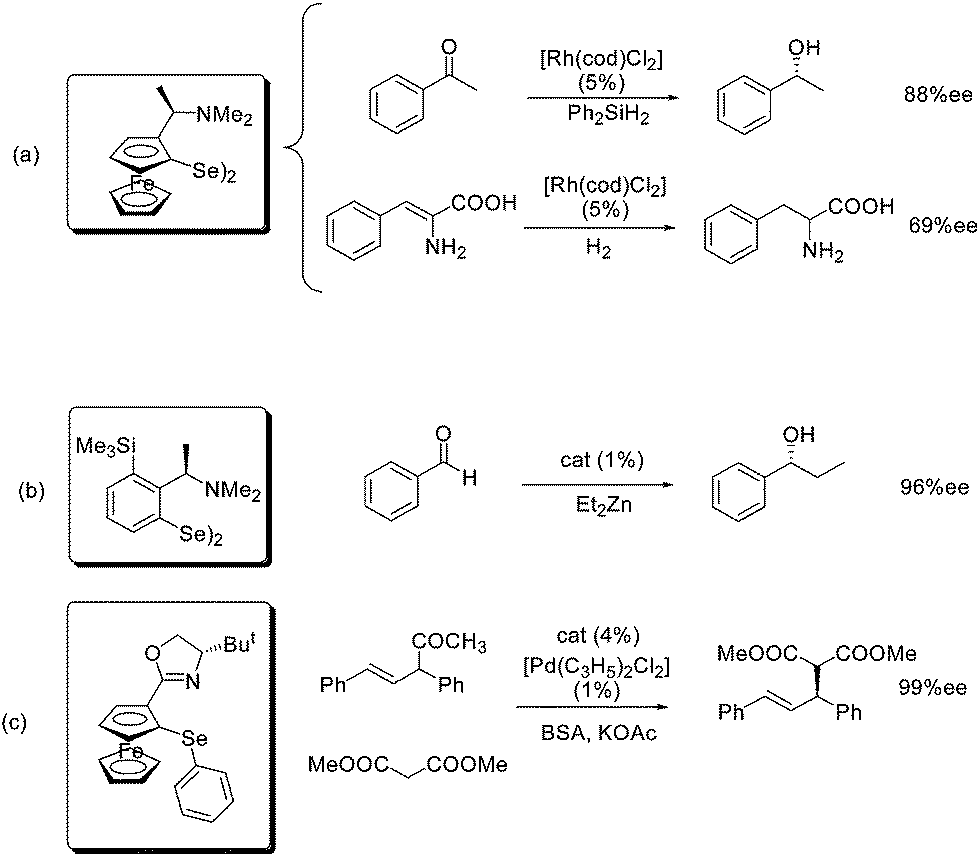 | ||
| Scheme 22 Organoselenium compounds in metal-catalyzed reactions. | ||
In all the cases the most favorable condition for the metal coordination is the presence of a nitrogen atom, with a selenium atom directly linked to an aromatic ring.
Palladium can be efficiently coordinated by a phenyl selenide derivative while zinc, rhodium ad other metals such as copper and tin require diselenides. These have been demonstrated, in almost all the cases, to be pre-catalysts of the actual catalytically active ligand–metal complexes.
4.2 One-pot selenation–deselention reactions
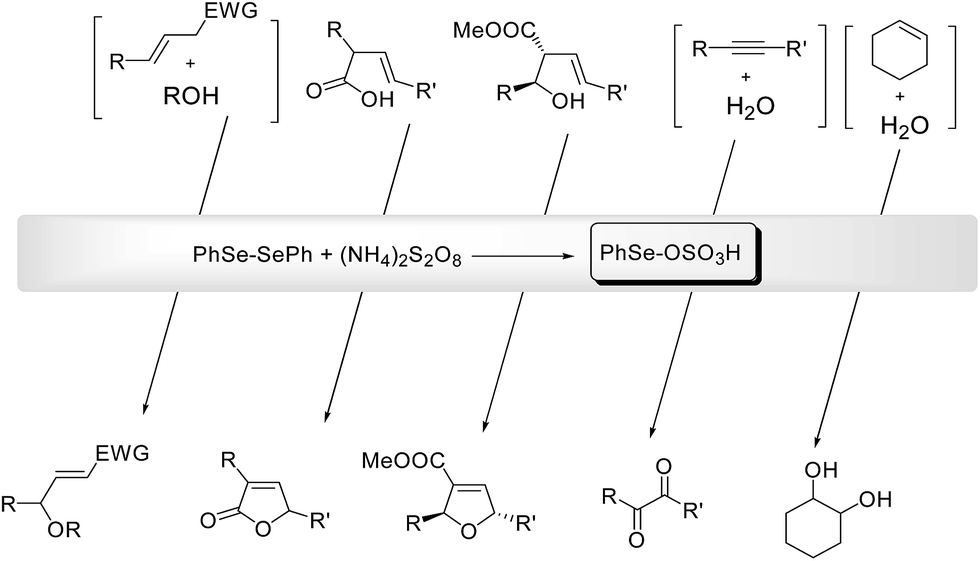
Electrophilic selenium reagents can be easily prepared from the corresponding diselenides by oxidation of the Se–Se bond. These reagents readily react with olefins and alkynes generating unstable seleniranium intermediates that can be opened by external or internal nucleophiles, leading to functionalized selenides through an addition or a cyclofunctionalization reaction, respectively.7aWhen the oxidant is ammonium persulfate the resulting selenide (see Scheme 23 as a general example of a cyclofunctionalization) can be further oxidized to the corresponding selenonium salt. This activates the selenium moiety towards elimination or substitution, depending on the reaction conditions. From these processes the electrophilic PhSe-sulfate, the actual catalyst of the process, is regenerated. In most cases diphenyl diselenide can be recovered at the end of the reaction and reused.
 | ||
| Scheme 23 One pot selenenylation–deselenenylation for the synthesis of heterocycles. | ||
The deselenation by elimination is favored by the presence of an electron-withdrawing group in a suitable position to stabilize, by conjugation, the new carbon–carbon double bond. Representative examples of one-pot catalytic reactions are reported in Scheme 24, including the synthesis of γ-methoxy- or γ-hydroxy-α,β-unsaturated esters and nitriles,55 the cyclization of alkenoic acids into butenolides56 and the highly stereoselective conversion of 2-cabomethoxy-3-alkenols into the corresponding 2,5-tetrahydrofurans.57
 | ||
| Scheme 24 Selected examples of one-pot catalytic conversions. | ||
When there are no structural conditions that can drive the reaction toward the elimination, and a suitable nucleophile is present (e.g. water as the solvent) a substitution of the selenonium moiety can occur. Using this procedure the oxidation of alkynes into the corresponding 1,2-diketo derivatives58 and the synthesis of vicinal diols59 have been efficiently achieved.
All the processes are the result of a stereospecific anti addition to a carbon–carbon double bond trough the formation of a seleniranium ion intermediate. Using catalytic amounts of optically pure diselenides some examples of asymmetric transformations were reported in moderate to good level of facial selectivity and using ammonium persulfate or PhI(OCOCF3)2 as oxidant.60
4.3 Organoselenium catalysts in oxygen transfer reactions
Oxidation reactions are among the most important processes for the chemical industry, but they often represent a problematic step in terms of waste production and use of toxic reagents. For these reasons the development of new catalysts able to activate “green” oxidants, such as oxygen or hydrogen peroxide, is a very active research area.It has been recently reported that the new bench-stable selenolate PhSeZnCl, in which selenium resembles the selenium atom in the enzyme Gpx (glutathione peroxidase), is able to promote the oxygen-mediated oxidation of thiols to the corresponding disulfides. Particularly interesting is the oxidation of glutathione and dithiothreitol that has been used to setup a simple and fast NMR assay for the evaluation of the GPx-like activity of organoselenium derivatives.61
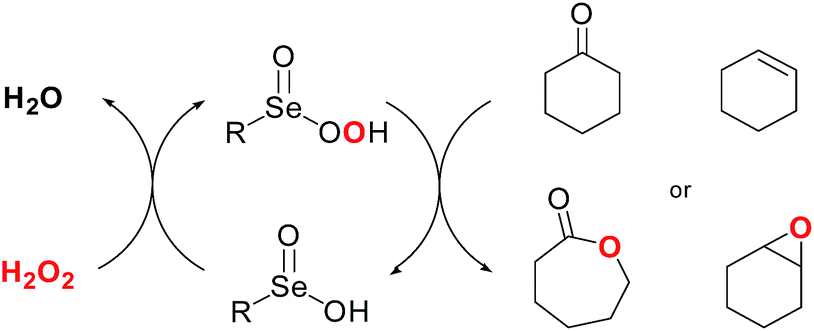
The use of organoselenium reagents for the activation of hydrogen peroxide was first reported for the Baeyer–Villiger reaction. In this case the actual oxygen transfer agent is a perseleninic acid. The catalytic efficiency is mainly dependent on the electronic nature of the selenium atom in the selenenic acid, and it is increased by the presence of electron-withdrawing groups such as nitro and trifluoromethyl moieties.62
The same catalysts, in the presence of hydrogen peroxide, promote the oxidation of carbon–carbon double bonds affording the corresponding epoxides in yield ranging from moderate to excellent (20–99%), when the reaction is performed in glycerol as recyclable reaction medium (Scheme 25).63
 | ||
| Scheme 25 Baeyer–Villiger and epoxidation reactions. | ||
When the epoxidation occurs in the presence of water as cosolvent, SeO2 and diphenyl diselenide catalyze the direct 1,2-dihydroxilation of olefins. It has been proposed that the mechanism involves a competition between an SN1 and SN2 ring opening reaction of the initially formed epoxide intermediate, and that the stereoselectivity depends both on the nature of the substrate as well as on the catalyst and the reaction conditions.64
The use of L-selenocysteine allows performing the reaction in water as the reaction medium, giving an enhanced catalyst turnover and a complete stereoselectivity in favor of the anti isomer. In addition, for endocyclic olefins, an interesting enantioselectivity has been obtained.65 From a sustainability point of view, it has been reported that the aqueous medium, which contains the water-soluble catalyst, can be reused without appreciable loss in yields for the first five reaction runs.
Very recently, the first example of an oxaziridinium catalyzed epoxidation reaction mediated by perselenenic acid has been reported. The mechanism involves a double catalytic cycle, in which the oxygen is transferred from hydrogen peroxide to the selenium and from selenium to an iminium ion with the formation of an oxaziridium intermediate that acts as the final catalyst. Chiral oxaziridinium 27 proved to be able to transfer the oxygen with an appreciable level of enantioselectivity (up to 85% ee) in the conversion of cyclic alkene 28 to epoxide 29 (Scheme 26).66
 | ||
| Scheme 26 Enantioselective epoxidation of olefins. | ||
Although yields and stereoselectivity are comparable with those obtained using Oxone® as stoichiometric oxidant (75% yield and 88% ee), the use of hydrogen peroxide reduces drastically the waste production affecting positively the atom economy of the process.
Perfluorooctylseleninic acid has been used for the allylic oxidation of alkenes to enones as well as for the oxidation of aryl ketones into the corresponding ketoacids. In this case the stoichiometric oxidant is iodoxybenzene and the catalyst can be completely recovered at the end of the reaction as diselenide.67
Organoselenium catalysts can also be used, with hydrogen peroxide as stoichiometric oxidant, for the oxidation of heteroatoms. Selenium derivative 30 has been recently used for the highly chemoselective oxidation of sulfide 31 to the corresponding sulfoxide 32 in the presence of trifluoroacetic acid (TFA) as cocatalyst.68 The authors demonstrated the presence of a peroxiselenurane intermediate as the actual catalyst of the oxidation reaction (Scheme 24). In the same paper the authors described the use of 30 as precatalyst in a number of different oxidation reactions. Interestingly, applying the reaction conditions described in Scheme 27, 1-methylcyclohexene can be converted into the corresponding epoxide in almost quantitative yield.
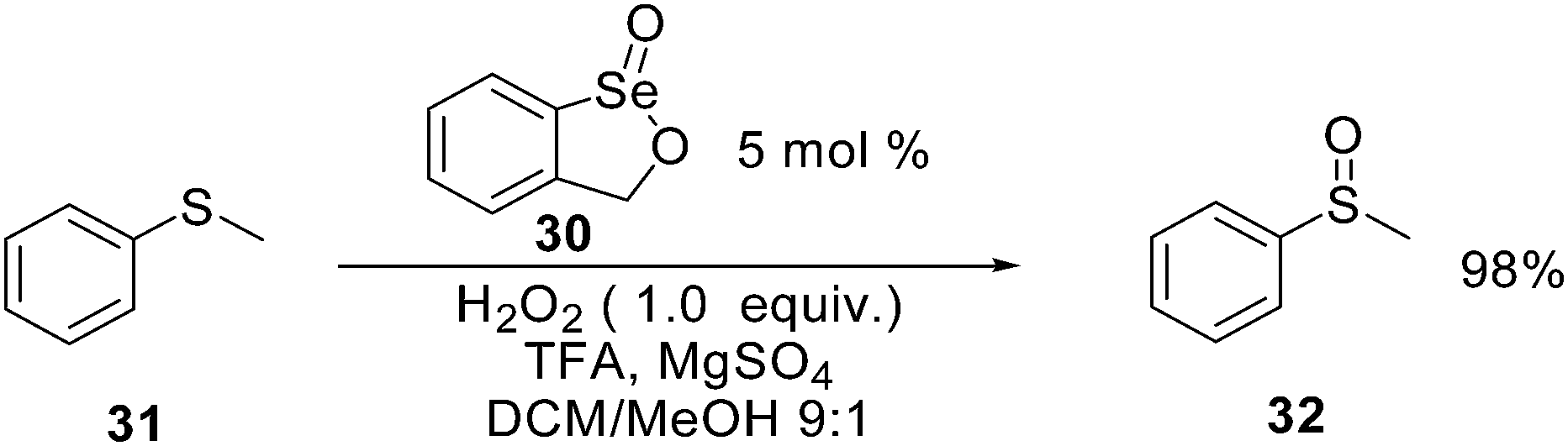 | ||
| Scheme 27 Oxidation of sulfides to sulfoxides. | ||
Similarly, using diphenyl diselenide in the presence of hydrogen peroxide, aromatic amines 33 can be oxidized to the corresponding nitroso derivatives 34. These could not be isolated but had to be trapped by reaction with a conjugated diene affording the product of a hetero-Diels–Alder reaction (Scheme 28).69
 | ||
| Scheme 28 Oxidation of arylamines. | ||
Very recently, it has been reported that similar reaction conditions promote the dehydration of aldoximes. The initially formed PhSeOH is readily converted into the corresponding seleninic anhydride PhSeOSePh (35) that, by reaction with substrate 36, affords the mixed anhydride 37. This undergoes a rearrangement to give the corresponding selenoxide 38 that, after a syn-elimination, leads to nitrile 39 (Scheme 29).70
 | ||
| Scheme 29 Aldoximes dehydration. | ||
5. Toxicology of selenium and selenium-containing compounds
Despite selenium being initially highlighted as a deadly poison, it has been later reinstated as an essential element because of its importance for living cells. Selenium toxicity was historically noted in 1285 when Marco Polo lost his horses crossing the Shanxi Province of China; this event was lately ascribed to the high concentration of this element in forages that caused selenosis in livestock. At the beginning of twentieth century, the chalcogen toxicity was experimentally demonstrated thanks to the seminal works by Franke,71 Painter,72 and Moxon73 who firstly defined the selenium poisoning. Nowadays, selenium is a well-established trace element with a recommended dietary intake of 55 μg for healthy adults per day in both USA and Europe and 50–250 μg per day for healthy Chinese adults.74 Selenium is also essential for many living organism such as bacteria, archea and less evolved eukaryotes.75 Even after the experimental demonstration of selenium importance for living systems, a sort of fear surrounded this element so much so that the term “selenophobia” was coined by Frost and Olson.76 The reason behind such perception relies in the narrow therapeutic window between beneficial amounts and toxic levels of some selenium derivatives.77 The upper limit of Se intake was indeed fixed by FDA and WHO at 400 μg per day,78 even if Poirer demonstrated that no adverse effects were observable with an amount of 853 μg per day.79The molecular mechanisms responsible for the toxicity of both organic and inorganic selenium species is not well understood, but the interaction with cellular thiols seems to play a central role. In addition, it was recently demonstrated that the oxidation of sulfhydryl groups could enhance the formation of reactive oxygen species (ROS).80
A lot of similarities as well as differences have been reported between the toxicity of inorganic and organic selenium species. Among inorganic derivatives, selenite (Se+4) and selenate (Se+6) are important in the biochemical cycle of the calchogen. They are enzymatically and/or non-enzymatically transformed into selenides (Se−2) that in turn are used as precursors for selenoprotein biosynthesis or eliminated through breathing or urination, after organication. It is really hard to find in the literature reliable information about toxic doses or hazardous levels of exposure for these inorganic species. On the other hand, many pharmaceutical applications have been claimed. For example, the dietary supplementation of 200 μg of Na2SeO3 enhanced the immunological response by improving the activity of T-lymphocytes,81 and, recently, a protective effect of this salt against mercury intoxication has been proven.82
Regarding organoselenium compounds, the situation is even more complex. This could possibly be a consequence of the more pronounced chemical diversity of organic Se-compounds, when compared to their inorganic counterparts. The metabolic fate of some organic derivatives somehow overlaps with that of inorganic ones. For example, it was demonstrated that selenocysteine and selenomethionine enzymatically generate Se−2.83 Selenomethionine is a natural source of Se for living organisms and exerts beneficial effects at 100–200 μg kg−1 in the diet of experimental animals, while toxicity is observable only far above (5 mg kg−1).84 In 2008 Johnson et al. performed a toxicological study on methylselenocysteine in the context of its potential use as breast cancer chemopreventive agent, observing hepatomegaly and a mild anemia in both rats and dogs at the highest tested dose, without observing any death.85
Besides naturally occurring organoselenium compounds, an increasing number of synthetic derivatives are appearing in literature reflecting the scientific interest for these compounds both in organic and medicinal chemistry. Selenium looks appealing from a medicinal chemistry point of view, given the relatively low number of Se-derivatives endowed with biological activities, but this underlines a gap of knowledge about their toxicity. The most biologically studied organoselenium compound is ebselen, which was evaluated in clinical trials for ischemic stroke,86 hearing loss,87 and recently for bipolar disorder.88 The toxicity of ebselen was studied by Nozawa, who intragastrically administered to mice 400 μg kg−1 of compound, reporting no toxic effects.89 More recently, the lethal dose (LD50) of a single intraperitoneal administration of ebselen was defined for rats (400 μg kg−1) and for mice (340 μg kg−1).90
Diphenyl diselenide is one of the most widely used organoselenium reagent in organic synthesis; it has multiple biological targets that called the attention of scientists but resulted in a toxicity picture that is still an unsolved puzzle.91 The LD50 was established in the same study of ebselen emerging as more tolerated in rats but less in mice, while no lethal effect was observed when the compound was administered orally92 or subcutaneously.93 In 2009 a series of substituted diaryl diselenides were evaluated under superimposable conditions indicating that the introduction of functional groups into aromatic rings did not increase the toxicity.94
Being more volatile than the other organoselenium compounds, some selenols are better known for their vile smell rather than for their toxicity or biological activity. In addition, they spontaneously turn into the corresponding diselenides thus showing the same biological profile of their dimeric counterparts.
In conclusion, a delineation of all selenium compounds as generally toxic is far from being realistic, since different selenium species, and different compounds within species, are capable of exerting diverse biological activities and thus show different degrees of toxicity.
Selenium waste handling is another matter of concern among organic chemists. Coming into contact with soils, organic and inorganic selenium species can be captured by plants.95 Among them, Indian mustard (Brassica juncea) and broccoli (B. oleracea) have been shown to be selenium hyperaccumulators.96 They are capable of decompose selenium derivatives transforming Se into the non-toxic and volatile dimethyl selenide.97 The detoxification of selenium by plants, a process termed phytoevaporation,98 may be utilized as an alternative strategy to remove the element from soils.
6. Conclusions
In this account we reviewed the field of organoselenium chemistry from the point of view of sustainability, focusing on both recent and historically relevant examples. All the processes described here can be related to some extent to the twelve principles of green chemistry, and can be ascribed into three main areas: the use of alternative solvents and/or reaction media, bio-inspired organoselenium catalysts and the use of non-conventional and energy-saving sources of activation. These represent, in our view, the principal aspects to consider in the development of more sustainable organic processes aimed at the synthesis of organoselenium compounds, or making use of selenium containing species as catalysts.A section discussing the toxicity of both inorganic and organic selenium-containing compounds is also included, with the hope that this could promote a more realistic view about the use of these compounds and their impact on the environment.
Acknowledgements
The authors kindly acknowledge CAPES for a fellowships to J.B.A (7706-13-8) and V.N (7569-13-0) and CINMPIS for a fellowship to L.S. C.S. also acknowledges RSC Advances for the Best Poster Award “Environmental & Green Chemistry” assigned during 12th Eurasia Conference on Chemical Sciences in Corfu, Greece.References
- (a) J. J. Berzelius, Afh. Fys., Kemi Mineral., 1818, 6, 42 Search PubMed; (b) K. B. Sharples, R. F. Law and A. Y. Teranishi, J. Am. Chem. Soc., 1973, 95, 6137 CrossRef.
- (a) K. C. Nicolau and N. A. Petasi, Selenium in Natural Products Synthesis, CIS, Philadelphia, PA, 1984 Search PubMed; (b) C. Paulmier, Selenium Reagents and Intermediates in Organic Synthesis, Pergamon, Oxford, U.K., 1986 Search PubMed; (c) S. Patai and Z. Rappoport, The Chemistry of Organic Selenium and Tellurium Compounds, Wiley, New York, 1986, vol. 1 Search PubMed; (d) D. Liotta, Organoselenium Chemistry, Wiley, New York, 1987 Search PubMed; (e) A. Krief and L. Hevesi, Organoselenium Chemistry I, Springer, Berlin, 1988 Search PubMed; (f) T. G. Back, Organoselenium Chemistry: A Practical Approach, Oxford University Press, Oxford, U.K., 1999 Search PubMed; (g) H. J. Reich, Acc. Chem. Res., 1979, 12, 22 CrossRef CAS; (h) D. Liotta, Acc. Chem. Res., 1984, 17, 28 CrossRef CAS; (i) T. Wirth, Organoselenium Chemistry – Modern Developments in Organic Synthesis, Topics in Current Chemistry 208, Spring-Verlag, Heidelberg, Germany, 2000 Search PubMed; (j) G. Mugesh and H. B. Singh, Acc. Chem. Res., 2002, 35, 226 CrossRef CAS PubMed.
- (a) G. Mugesh and H. Singh, Chem. Soc. Rev., 2000, 29, 347 RSC; (b) G. Mugesh, W. W. Du Mont and H. Sies, Chem. Rev., 2001, 101, 2125 CrossRef CAS PubMed; (c) C. W. Nogueira, G. Zeni and J. B. T. Rocha, Chem. Rev., 2004, 104, 6255 CrossRef CAS PubMed; (d) B. K. Sarma and G. Mugesh, Org. Biomol. Chem., 2008, 6, 965–974 RSC; (e) E. E. Alberto, V. Nascimento and A. L. Braga, J. Braz. Chem. Soc., 2010, 21, 2032 CrossRef CAS PubMed; (f) C. T. Santi, C. Tidei, C. Scalera, M. Piroddi and F. Galli, Curr. Chem. Biol., 2013, 7, 25 CrossRef CAS; (g) E. E. Alberto and A. L. Braga, Selenium and Tellurium Chemistry – From Small Molecules to Biomolecules and Materials, ed. W. J. Derek and L. Risto, Springer-Verlag, Berlin, 2011 Search PubMed; (h) A. L. Braga and J. Rafique, Synthesis of biologically relevant small molecules containing selenium. Part A. Antioxidant compounds, The Chemistry of Organic Selenium and Tellurium Compounds, ed. Z. Rappoport, John Wiley & Sons, Ltd, Chichester, UK, 2013, vol. 4 Search PubMed; (i) A. L. Braga and J. Rafique, Synthesis of biologically relevant small molecules containing selenium. Part B. Anti-infective and anticancer Compounds, The Chemistry of Organic Selenium and Tellurium Compounds, ed. Z. Rappoport, John Wiley & Sons, Ltd, Chichester, UK, 2013, vol. 4 Search PubMed; (j) A. L. Braga and J. Rafique, Synthesis of biologically relevant small molecules containing selenium. Part C. Miscellaneous biological activities, The Chemistry of Organic Selenium and Tellurium Compounds, ed. Z. Rappoport, John Wiley & Sons, Ltd, Chichester, UK, 2013, vol. 4 Search PubMed; (k) C. Jacob, G. I. Giles, N. M. Giles and H. Sies, Angew. Chem., Int. Ed., 2003, 42, 4742–4758 CrossRef CAS PubMed.
- (a) Benign by Design: Alternative Synthetic Design for Pollution Prevention, ed. P. Anastas and C. Farris, American Chemical Society, Washington, DC, 1994 Search PubMed; (b) I. T. Horváth and P. T. Anastas, Chem. Rev., 2007, 2167 Search PubMed.
- (a) World Comission on Environment and Development, Our Common Future, Oxford University Press, New York, 1987 Search PubMed; (b) United Nations Environment Programme: Environment for Development, http://www.unep.org/Documents.Multilingual/Default.asp?documentid=97%26articleid=1503, accessed January 19, 2014; (c) P. T. Anastas and J. C. Warner, Green Chemistry: Theory and Practice, University Press, USA, 2000 Search PubMed.
- D. M. Freudendahl, S. Santoro, S. A. Shahzad, C. Santi and T. Wirth, Angew. Chem., Int. Ed., 2009, 48, 8409 CrossRef CAS PubMed.
- (a) C. Santi and C. Tidei, Electrophilic selenium/tellurium reagents: Reactivity and their contribution to Green Chemistry, The Chemistry of Organic Selenium and Tellurium Compounds, ed. Z. Rappoport, John Wiley & Sons, Ltd, Chichester, UK, 2013, vol. 4 Search PubMed; (b) M. Godoi, D. G. Liz, E. W. Ricardo, M. S. T. Rocha, J. B. Azeredo and A. L. Braga, Tetrahedron, 2013, 71, 1 Search PubMed.
- (a) A. Chanda and V. V. Fokin, Chem. Rev., 2009, 109, 725 CrossRef CAS PubMed; (b) U. M. Lindstrom, Chem. Rev., 2002, 202, 2751 CrossRef PubMed.
- (a) A. V. Kumar, V. P. Reddy, C. S. Reddy and K. R. Rao, Tetrahedron Lett., 2011, 52, 3978 CrossRef CAS PubMed; (b) R. Sridhar, B. Srinivas, K. Surendra, N. S. Krishnaveni and K. R. Rao, Tetrahedron Lett., 2005, 46, 8837 CrossRef CAS PubMed.
- C. Tidei, Synlett, 2012, 23, 2865 CrossRef PubMed.
- (a) C. Santi, S. Santoro, B. Battistelli, L. Testaferri and M. Tiecco, Eur. J. Org. Chem., 2008, 5287 Search PubMed; (b) C. Santi, Phenylselenenylzinc halides, Encyclopedia of reagents for organic synthesis, ed. L. A. Paquette, John Wiley & Sons, New York, 2011 Search PubMed; (c) C. Tidei, M. Piroddi, F. Galli and C. Santi, Tetrahedron Lett., 2012, 53, 232 CrossRef CAS PubMed; (d) B. Battistelli, L. Testaferri, M. Tiecco and C. Santi, Eur. J. Org. Chem., 2011, 1848 CrossRef CAS; (e) S. Santoro, B. Battistelli, L. Testaferri, M. Tiecco and C. Santi, Eur. J. Org. Chem., 2009, 4921 CrossRef CAS.
- S. Narayan, J. Muldoon, M. G. Finn, V. V. Fokin, H. C. Kolb and K. B. Sharpless, Angew. Chem., Int. Ed., 2005, 44, 3275 CrossRef CAS PubMed.
- C. Santi, B. Battistelli, L. Testaferri and M. Tiecco, Green Chem., 2012, 14, 1277 RSC.
- A. Saha, D. Saha and B. C. Ranu, Org. Biomol. Chem., 2009, 7, 1652 CAS.
- (a) Z. U. Wang, Y.-S. Wang, P.-J. Pai, W. K. Russell, D. H. Russell and W. R. Liu, Biochemistry, 2012, 51, 5232–5234 CrossRef CAS PubMed; (b) J. Guo, J. Wang, J. Seok Lee and P. G. Schultz, Angew. Chem., Int. Ed., 2008, 47, 6399–6401 CrossRef CAS PubMed.
- S. D. Townsend, Z. Tan, S. Dong, S. Shang, J. A. Brailsford and S. J. Danishefsky, J. Am. Chem. Soc., 2012, 134, 3912–3916 CrossRef CAS PubMed.
- Y. A. Lin, J. M. Chalker and B. G. Davis, J. Am. Chem. Soc., 2010, 132, 16805–16811 CrossRef CAS PubMed.
- L. R. Malins and R. J. Payne, Org. Lett., 2012, 14, 3142–3145 CrossRef CAS PubMed.
- Y. A. Lin, O. Boutureira, L. Lercher, B. Bhushan, R. S. Paton and B. G. Davis, J. Am. Chem. Soc., 2013, 135, 12156–12159 CrossRef CAS PubMed.
- R. M. Hodlur and M. K. Rabinal, Chem. Eng. J., 2014, 244, 82–88 CrossRef CAS PubMed.
- For reviews, see: (a) J. Dupont, R. F. Souza and P. A. Z. Suarez, Chem. Rev., 2002, 102, 3667 CrossRef CAS PubMed; (b) M. E. Zakrzewska, E. B. Lukasik and R. B. Lukasik, Chem. Rev., 2011, 111, 397 CrossRef CAS PubMed; (c) J. P. Hallett and T. Welton, Chem. Rev., 2011, 111, 3508 CrossRef CAS PubMed.
- (a) G. Tabarelli, E. E. Alberto, A. M. Deobald, G. Marin, O. E. D. Rodrigues, L. Dornelles and A. L. Braga, Tetrahedron Lett., 2010, 51, 5728 CrossRef CAS PubMed; (b) S. Narayanaperumal, K. Gul, C. Kawasoko, D. Singh, L. Dornelles, O. E. D. Rodrigues and A. L. Braga, J. Braz. Chem. Soc., 2010, 21, 2079 CrossRef CAS PubMed; (c) S. Narayanaperumal, E. E. Alberto, F. M. de Andrade, E. J. Lenardão, P. S. Taube and A. L. Braga, Org. Biomol. Chem., 2009, 7, 4647 RSC; (d) K. Gul, S. Narayanaperumal, L. Dornelles, O. E. D. Rodrigues and A. L. Braga, Tetrahedron Lett., 2011, 52, 3592 CrossRef CAS PubMed; (e) H. Zhao, W. Hao, Z. Xi and M. Cai, New J. Chem., 2011, 35, 2661 RSC; (f) S. Banerjee, L. Adak and B. C. Ranu, Tetrahedron Lett., 2012, 53, 2149 CrossRef CAS PubMed; (g) E. J. Lenardão, L. G. Dutra, M. T. Saraiva, R. G. Jacob and G. Perin, Tetrahedron Lett., 2007, 48, 8011 CrossRef PubMed; (h) M.-H. Yang, G.-B. Yan and Y.-F. Zheng, Tetrahedron Lett., 2008, 49, 6471 CrossRef CAS PubMed; (i) J. Vargas, S. Narayanaperumal, K. Gul, B. Ravanello, L. Dornelles, L. C. Soares, C. F. S. Alves, T. Schneider, R. Vaucher, R. C. Santos and O. E. D. Rodrigues, Tetrahedron, 2012, 68, 10444 CrossRef CAS PubMed; (j) S. M. Salman, R. S. Schwab, E. E. Alberto, J. Vargas, L. Dornelles, O. E. D. Rodrigues and A. L. Braga, Synlett, 2011, 69 CAS; (k) S. M. Salman, S. Narayanaperumal, R. S. Schwab, C. R. Bender, O. E. D. Rodrigues and L. Dornelles, RSC Adv., 2012, 2, 8478 RSC.
- D. Singh, S. Narayanaperumal, K. Gul, M. Godoi, O. E. D. Rodrigues and A. L. Braga, Green Chem., 2010, 12, 957 RSC.
- S. Narayanaperumal, E. E. Alberto, K. Gul, C. Y. Kawasoko, L. Dornelles, O. E. D. Rodrigues and A. L. Braga, Tetrahedron, 2011, 67, 4723 CrossRef CAS PubMed.
- S. Narayanaperumal, E. E. Alberto, K. Gul, O. E. D. Rodrigues and A. L. Braga, J. Org. Chem., 2010, 75, 3886 CrossRef CAS PubMed.
- D. Singh, E. E. Alberto, O. E. D. Rodrigues and A. L. Braga, Green Chem., 2009, 11, 1521 RSC.
- C. S. Freitas, A. M. Barcellos, V. G. Ricordi, J. M. Pena, G. Perin, R. G. Jacob, E. J. Lenardão and D. Alves, Green Chem., 2011, 13, 2931 RSC.
- (a) F. W. Lichtenthaler and S. Peters, C. R. Chim., 2004, 7, 65 CrossRef CAS PubMed; (b) Y. Zheng, X. Chen and Y. Shen, Chem. Rev., 2008, 108, 5253 CrossRef CAS PubMed.
- D. T. Johnson and K. A. Taconi, Environ. Prog., 2007, 26, 338 CrossRef CAS.
- Y. Gu and F. Jèrôme, Green Chem., 2010, 12, 1127 RSC.
- (a) V. G. Ricordi, C. S. Freitas, G. Perin, E. J. Lenardão, R. G. Jacob, L. Savegnago and D. Alves, Green Chem., 2012, 14, 1030 RSC; (b) L. C. Gonçalves, G. F. Fiss, G. Perin, D. Alves, R. G. Jacob and E. J. Lenardão, Tetrahedron Lett., 2010, 51, 6772 CrossRef PubMed.
- F. N. Victoria, C. S. Radatz, M. Sachini, R. G. Jacob, G. Perin, W. P. da Silva and E. J. Lenardão, Tetrahedron Lett., 2009, 50, 6761 CrossRef CAS PubMed.
- (a) R. S. Varma, Green Chem., 1999, 1, 43 RSC; (b) K. Tanaka and F. Toda, Chem. Rev., 2000, 100, 1025 CrossRef CAS PubMed; (c) P. J. Walsh, H. Li and C. A. de Parrodi, Chem. Rev., 2007, 107, 2503 CrossRef CAS PubMed.
- (a) M. Godoi, E. W. Ricardo, G. V. Botteselle, F. Z. Galetto, J. B. Azeredo and A. L. Braga, Green Chem., 2012, 14, 456 RSC; (b) G. Perin, R. G. Jacob, L. G. Dutra, F. Azambuja, G. F. Santos and E. J. Lenardão, Tetrahedron Lett., 2006, 47, 935 CrossRef CAS PubMed; (c) G. Perin, S. R. Mendes, M. S. Silva, E. J. Lenardão, R. G. Jacob and P. C. Santos, Synth. Commun., 2006, 36, 2587 CrossRef CAS; (d) N. Mukherjee, T. Chatterjee and B. C. Ranu, J. Org. Chem., 2013, 78, 11110 CrossRef CAS PubMed.
- V. P. Ananikov and I. P. Beletskaya, Org. Biomol. Chem., 2004, 2, 284 CAS.
- G. Marin, A. L. Braga, A. S. Rosa, F. Z. Galetto, R. A. Burrow, H. Gallardo and M. W. Paixão, Tetrahedron, 2009, 65, 4614 CrossRef CAS PubMed.
- (a) S. A. Galema, Chem. Soc. Rev., 1997, 26, 233 RSC; (b) D. Adam, Nature, 2003, 421, 571 CrossRef CAS PubMed; (c) C. O. Kappe, Angew. Chem., Int. Ed., 2004, 42, 6250 CrossRef PubMed; (d) D. Dallinger and C. O. Kappe, Chem. Rev., 2007, 107, 2563 CrossRef CAS PubMed; (e) V. Polshettiwar and R. S. Varma, Acc. Chem. Res., 2008, 41, 629 CrossRef CAS PubMed; (f) S. Ravichandran and E. Karthikeyan, Int. J. ChemTech Res., 2011, 3, 466 Search PubMed; (g) M. Godoi, G. V. Botteselle, J. Rafique, M. S. T. Rocha, J. M. Pena and A. L. Braga, Asian J. Org. Chem., 2013, 2, 746 CrossRef CAS; (h) J. B. Azeredo, M. Godoi, R. S. Schwab, G. V. Botteselle and A. L. Braga, Eur. J. Org. Chem., 2013, 5188 CrossRef CAS.
- J. D. Moseley and C. O. Kappe, Green Chem., 2011, 13, 794 RSC.
- D. Obermayer, B. Gutmann and C. O. Kappe, Angew. Chem., Int. Ed., 2009, 48, 1 CrossRef CAS PubMed For a review, see: C. O. Kappe and D. Dallinger, Mol. Diversity, 2009, 13, 71 CrossRef PubMed.
- (a) G. Perin, R. G. Guimarães, F. de Azambuja, G. F. F. dos Santos and E. J. Lenardão, Tetrahedron Lett., 2006, 47, 935 CrossRef CAS PubMed; (b) G. Perin, S. R. Mendes, M. S. Silva, E. J. Lenardão, R. G. Guimarães and P. C. S. Santos, Synth. Commun., 2006, 18, 2587 CrossRef; (c) G. Perin, R. G. Jacob, F. de Azambuja, G. V. Botteselle, G. M. Siqueira, R. A. Freitag and E. J. Lenardão, Tetrahedron Lett., 2005, 46, 1679 CrossRef CAS PubMed.
- S. Kumar and L. Engman, J. Org. Chem., 2006, 71, 5400 CrossRef CAS PubMed.
- D. Kundu, S. Ahammed and B. C. Ranu, Green Chem., 2012, 14, 2024 RSC.
- G. V. Botteselle, M. Godoi, F. Z. Galetto, L. Bettanin, D. Singh, O. E. D. Rodrigues and A. L. Braga, J. Mol. Catal. A: Chem., 2012, 365, 186 CrossRef CAS PubMed.
- A. L. Braga, F. Vargas, J. A. Sehnem and L. A. Wessjohann, Eur. J. Org. Chem., 2006, 4993 CrossRef CAS.
- J. B. Azeredo, M. Godoi, G. M. Martins, C. C. Silveira and A. L. Braga, J. Org. Chem., 2014, 79, 4125 CrossRef CAS PubMed.
- For reviews see: (a) N. Baig and R. S. Varma, Chem. Soc. Rev., 2012, 41, 1559 RSC; (b) R. Cella and H. A. Stefani, Tetrahedron, 2009, 65, 2619 CrossRef CAS PubMed , Ver refrencias artigo do Ricardo 19.
- (a) P. R. Gogate and A. B. Pandit, Ultrason. Sonochem., 2003, 10, 325 CrossRef CAS; (b) M. Ashokkumar, J. Lee, S. Kentish and F. Grieser, Ultrason. Sonochem., 2007, 14, 470 CrossRef CAS PubMed.
- (a) T. Cheng, X. Zheng and Q. Ke, J. Chem. Res., 2011, 522 CrossRef CAS; (b) B. Gautheron, G. Tainturier and C. Degrand, J. Am. Chem. Soc., 1985, 107, 5579 CrossRef CAS; (c) D. P. Thompson and P. Boudjouk, J. Org. Chem., 1988, 53, 2109 CrossRef CAS; (d) F. V. Singh and T. Wirth, Org. Lett., 2011, 13, 6504 CrossRef CAS PubMed.
- (a) R. Vukievic, S. Konstantinovic and M. L. Mihailovic, Tetrahedron, 1991, 47, 859 CrossRef.
- C. Santi, S. Santoro and B. Battistelli, Curr. Org. Chem., 2010, 14, 2442–2462 CrossRef CAS.
- N. Sonoda, G. Yamamoto, K. Natsukawa, K. Kondo and S. Murai, Tetrahedron Lett., 1975, 24, 1969–1972 CrossRef.
- (a) Y. Nishibayashy, J. D. Singh, J. Arikawa, S. Uemura and M. Hidai, Organomet. Chem., 1997, 531, 13–18 CrossRef; (b) C. Borner, M. R. Dennis, E. Sinn and S. Woodward, Eur. J. Org. Chem., 2001, 2435–2446 CrossRef CAS.
- C. Santi and T. Wirth, Tetrahedron: Asymmetry, 1999, 10, 1019–1023 CrossRef CAS.
- S.-L. You, X.-L. Hou and L.-X. Dai, Tetrahedron: Asymmetry, 2000, 11, 1495–1500 CrossRef CAS.
- M. Tiecco, L. Testaferri, M. Tingoli, L. Bagnoli and C. Santi, J. Chem. Soc., Chem. Commun., 1993, 637–639 RSC.
- M. Tiecco, L. Testaferri, M. Tingoli, L. Bagnoli and C. Santi, Synlett, 1993, 798–800 CrossRef CAS PubMed.
- M. Tiecco, L. Testaferri and C. Santi, Eur. J. Org. Chem., 1999, 797–803 CrossRef CAS.
- S. Santoro, B. Battistelli, B. Gjoka, C. W. S. Si, L. Testaferri, M. Tiecco and C. Santi, Synlett, 2010, 1402–1406 CAS.
- C. Santi, M. Tiecco, L. Testaferri, C. Tomassini, S. Santoro and G. Bizzoca, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 956–960 CrossRef CAS.
- (a) D. M. Browne, O. Nyiomura and T. Wirth, Phosphorus, Sulfur Silicon Relat. Elem., 2008, 183, 1026–1035 CrossRef; (b) D. M. Browne, O. Nyiomura and T. Wirth, Org. Lett., 2007, 9, 3169–3171 CrossRef CAS PubMed.
- C. Tidei, F. Piroddi, F. Galli and C. Santi, Tetrahedron Lett., 2012, 53, 232–234 CrossRef CAS PubMed.
- (a) P. A. Grieco, Y. Yokoyama, S. Gilman and Y. Ohfune, J. Chem. Soc., Chem. Commun., 1977, 870–871 RSC; (b) L. Syper and J. Mlochowski, Tetrahedron, 1987, 43, 207–213 CrossRef CAS; (c) L. Syper, Tetrahedron, 1987, 43, 2853–2871 CrossRef CAS; (d) L. Syper, Synthesis, 1989, 167–172 CrossRef CAS; (e) G.-J. ten Brink, J.-M. Vis, I. W. E. Arends and R. A. Sheldon, J. Org. Chem., 2001, 66, 2429–2433 CrossRef CAS PubMed.
- H. Garcia Marin, J. C. van der Toorn, J. A. Myoral, J. I. Garcia and I. W. C. E. Arends, Green Chem., 2009, 11, 1605–1609 RSC.
- S. Santoro, C. Santi, M. Sabatini, L. Testaferri and M. Tiecco, Adv. Synth. Catal., 2008, 350, 2881–2884 CrossRef CAS.
- C. Santi, R. Di Lorenzo, C. Tidei, L. Bagnoli and T. Wirth, Tetrahedron, 2012, 68, 10530–10535 CrossRef CAS PubMed.
- B. R. Buckley, C. E. Elliott, Y. Chan, N. Dreyfus and P. C. Bulman Page, Synlett, 2013, 24, 2266–2270 CrossRef CAS PubMed.
- (a) D. Crich and Y. Zou, Org. Lett., 2004, 6, 775–777 CrossRef CAS PubMed; (b) D. Crich and Y. Zou, J. Org. Chem., 2005, 70, 163309–163311 Search PubMed.
- E. A. Mercier, C. D. Smith, M. Parvez and T. G. Back, J. Org. Chem., 2012, 77, 3508–3517 CrossRef CAS PubMed.
- D. Zhao, M. Johanson and J.-E. Bäckvall, J. Org. Chem., 2007, 72, 4431–4436 CrossRef PubMed.
- L. Yu, H. Li, X. Zhang, J. Ye, J. Liu, Q. Xu and M. Lautens, Org. Lett., 2014, 16, 1346–1349 CrossRef CAS PubMed.
- (a) K. D. Franke, J. Nutr., 1934, 8, 597–608 CAS; (b) K. D. Franke, J. Nutr., 1934, 8, 609–613 CAS.
- E. P. Painter, Chem. Rev., 1941, 28, 179–213 CrossRef CAS.
- A. L. Moxon and M. Rhian, Physiol. Rev., 1943, 23, 305–337 CAS.
- H. J. Sun, B. Rathinasabapathi, B. Wu, J. Luo, L. P. Pu and L. Q. Ma, Environ. Int., 2014, 69C, 148–158 CrossRef PubMed.
- A. V. Lobanov, D. L. Hatfield and V. N. Gladyshev, Biochim. Biophys. Acta, 2009, 11, 1424–1428 CrossRef PubMed.
- D. V. Frost and O. E. Olson, CRC Crit. Rev. Toxicol., 1972, 4, 467–514 CrossRef.
- D. Taylor, C. Dalton, A. Hall, M. N. Woodroofe and P. H. Gardiner, Br. J. Biomed. Sci., 2009, 66, 107–116 CAS.
- E. R. Monsen, J. Am. Diet. Assoc., 2000, 100, 637–640 CrossRef CAS.
- Y. Ogra and Y. Anan, J. Anal. At. Spectrom., 2009, 24, 1477–1488 RSC.
- (a) D. Plano, Y. Baquedano, E. Ibanez, I. Jimenez, J. A. Palop, J. E. Spallholz and C. Sanmartin, Molecules, 2010, 15, 7292–7312 CrossRef CAS PubMed; (b) J. J. Chen, L. M. Boylan, C. K. Wu and J. E. Spallholz, BioFactors, 2007, 31, 55–66 CrossRef CAS.
- L. Kiremidjian-Schumacher, M. Roy, H. I. Wishe, M. W. Cohen and G. Stotzky, Biol. Trace Elem. Res., 1994, 41, 115–127 CrossRef CAS.
- (a) S. Kalender, F. G. Uzun, F. Demir, M. Uzunhisarcikli and A. Aslanturk, Food Chem. Toxicol., 2013, 55, 456–462 CrossRef CAS PubMed; (b) V. Branco, J. Canario, J. Lu, A. Holmgren and C. Carvalho, Free Radical Biol. Med., 2012, 52, 781–793 CrossRef CAS PubMed.
- (a) K. T. Suzuki, K. Kurasaki and N. Suzuki, Biochim. Biophys. Acta, Gen. Subj., 2007, 1770, 1053–1061 CrossRef CAS PubMed; (b) Y. Ohta and K. T. Suzuki, Toxicol. Appl. Pharmacol., 2008, 226, 169–177 CrossRef CAS PubMed.
- M. Jacobs and C. Forst, J. Toxicol. Environ. Health, 1981, 8, 575–585 CrossRef CAS PubMed.
- W. D. Johnson, R. L. Morrissey, I. Kapetanovic, J. A. Crowell and D. L. McCormick, Food Chem. Toxicol., 2008, 46, 1068–1078 CrossRef CAS PubMed.
- T. Yamaguchi, K. Sano, K. Takakura, I. Saito, Y. Shinohara, T. Asano and H. Yasuhara, Journal of Cerebral Circulation, 1998, 29, 12–17 CrossRef CAS.
- J. Kil, C. Pierce, H. Tran, R. Gu and E. D. Lynch, Hear. Res., 2007, 226, 44–51 CrossRef CAS PubMed.
- N. Singh, A. C. Halliday, J. M. Thomas, O. V. Kuznetsova, R. Baldwin, E. C. Woon, P. K. Aley, I. Antoniadou, T. Sharp, S. R. Vasudevan and G. C. Churchill, Nat. Commun., 2013, 4, 1332 CrossRef PubMed.
- R. Nozawa, M. Arai, R. Kuruto, T. Motohashi and H. Masayasu, J. Pharm. Pharmacol., 1996, 48, 64–67 CrossRef CAS PubMed.
- F. C. Meotti, V. C. Borges, G. Zeni, J. B. Rocha and C. W. Nogueira, Toxicol. Lett., 2003, 143, 9–16 CrossRef CAS.
- C. W. Nogueira and J. B. Rocha, Arch. Toxicol., 2011, 85, 1313–1359 CrossRef CAS PubMed.
- E. A. Wilhelm, C. R. Jesse, M. R. Leite and C. W. Nogueira, Pathophysiology, 2009, 16, 31–37 CrossRef CAS PubMed.
- C. W. Nogueira, E. B. Quinhones, E. A. Jung, G. Zeni and J. B. Rocha, Inflammation Res., 2003, 52, 56–63 CrossRef CAS.
- L. Savegnago, C. R. Jesse and C. W. Nogueira, Environ. Toxicol. Pharmacol., 2009, 27, 271–276 CrossRef CAS PubMed.
- D. R. Ellis and D. E. Salt, Curr. Opin. Plant Biol., 2003, 6, 273–279 CrossRef CAS.
- E. Dumont, F. Vanhaecke and R. Cornelis, Anal. Bioanal. Chem., 2006, 385, 1304–1323 CrossRef CAS PubMed.
- A. Zayed, C. M. Lytle and N. Terry, Planta, 1998, 206, 284–292 CrossRef CAS.
- M. Bodnar, P. Konieczka and J. Namiesnik, J. Environ. Sci. Health, Part C: Environ. Carcinog. Ecotoxicol. Rev., 2012, 30, 225–252 CAS.
| This journal is © The Royal Society of Chemistry 2014 |