 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Novel porous materials based on oligospiroketals (OSK)†
Pablo
Wessig
*a,
Maik
Gerngroß
a,
Simon
Pape
a,
Philipp
Bruhns
a and
Jens
Weber‡
b
aInstitut für Chemie, Universität Potsdam, Karl-Liebknecht-Str. 24-25, 14476 Potsdam, Germany. E-mail: wessig@uni-potsdam.de; Fax: +49-331977-5065; Tel: +49-331977-5401
bDepartment of Colloid Chemistry, Max Planck Institute of Colloids and Interfaces, Research Campus Golm, D-14476 Potsdam, Germany
First published on 3rd July 2014
Abstract
New porous materials based on covalently connected monomers are presented. The key step of the synthesis is an acetalisation reaction. In previous years we used acetalisation reactions extensively to build up various molecular rods. Based on this approach, investigations towards porous polymeric materials were conducted by us. Here we wish to present the results of these studies in the synthesis of 1D polyacetals and porous 3D polyacetals. By scrambling experiments with 1D acetals we could prove that exchange reactions occur between different building blocks (evidenced by MALDI-TOF mass spectrometry). Based on these results we synthesized porous 3D polyacetals under the same mild conditions.
Introduction
Porous materials have attracted the attention of scientists due their manifold uses in various technologies such as separation or catalysis. A high specific surface area is often beneficial and is realized especially in microporous materials, i.e. materials having pore sizes below 2 nm. Traditional examples are zeolites and activated carbons.1 Metal–organic frameworks (MOF) or porous coordination polymers (PCP) have been introduced by various research groups during the last years as additions to the classic materials.2–4 Most recently, interest in purely organic, but well-defined microporous materials arose and microporous organic polymers (MOP) have been developed following various concepts such as hypercrosslinking, intrinsic microporosity.5–12 Besides these amorphous materials, highly porous crystalline covalent organic frameworks (COF) and low-molecular weight cages have also been reported.13,14A common feature of all microporous polymers is the necessity of highly rigid strands and chains in order to prevent pore collapse. To ensure high microporosity in the final materials they need to feature either regular sites of contortion (such as a spirocenter or a tetraphenyl methane building block) to prevent dense chain packing or they need to be synthesized under well- controlled phase separation conditions (porogenic solvent) in order to fix the loose structure instead of forming dense polymer phases.
In the geometric sense, many of the above mentioned organic frameworks could be imagined to be composed of more or less long stiff molecular rods. Such molecules are characterized by a high aspect ratio and conformational rigidity and are the object of intense research efforts. Numerous applications both in life- and material-sciences were indeed already developed, which were summarized in some reviews.15 Basically, molecular rods may be subdivided in conjugated and non-conjugated rods. Whereas the former are important for organic electronics, the latter are predestined for biological and biochemical applications. Some years ago we developed a new class of non-conjugated molecular rods whose key elements are ketal structures. We therefore called these rods oligospiroketal (OSK) rods.16–22 The general structure of OSK rods (A) is depicted in Fig. 1. The backbone of the rods consists of spirocyclically joined 1,3-dioxane and cyclohexane rings, which are formed by ketalization of cyclohexan-1,4-dione E (or synthetic equivalents) with pentaerythritol B. Terminal functionalities are introduced by piperidine-4-one building blocks D. Furthermore, solubility enhancing building blocks (so-called “sleeves”, C) are often used due to the scarce solubility of longer OSK rods (Fig. 1).17,20 Meanwhile, we developed a series of mainly biochemical applications of OSK rods.18,19,21,22
 | ||
| Fig. 1 General structure of OSK rods (A) and building blocks (B–E). | ||
Building on the experience with OSK rods and their structural similarity to the basic requirements of microporous polymers, we report here on some novel porous materials with polyacetals (PA) structure whose common structural element is the 2,4,8,10-tetraoxaspiro[5.5]undecane (TOSU) skeleton. Therefore we call these structures TOSU-PA. The synthesis of the rods is analyzed in detail before the synthesis of microporous structures and their characterization by means of gas adsorption is discussed.
Results and discussion
The synthesis of TOSU-polyacetals relies on the acetalization method developed by Noyori.23 The bis-trimethylsilyl ethers of 1,2- and 1,3-diols react with aldehydes or ketones, catalyzed by trimethylsilyl trifluoromethanesulfonate (TMS-OTf), under very mild conditions to yield the corresponding acetals. A special feature of this method is that the reactants can be used in equimolar amounts in contrast to the “classic” method, which involves normally an excess of one of the reactants. The key reagent for the preparation of TOSU-polyacetals is the tetrakis-trimethylsilyl ether of pentaerythritol 2.24 This compound was treated with five different carbonyl compounds 1a–e with varying number of carbonyl groups. The synthetic route to TOSU-polyacetals TOSU-PA-a–e and the structure of building blocks 1a–e is summarized in Scheme 1. Depending on the number of carbonyl groups we expect linear 1D (1a, b) and cross-linked (1c, d, e) polyacetals.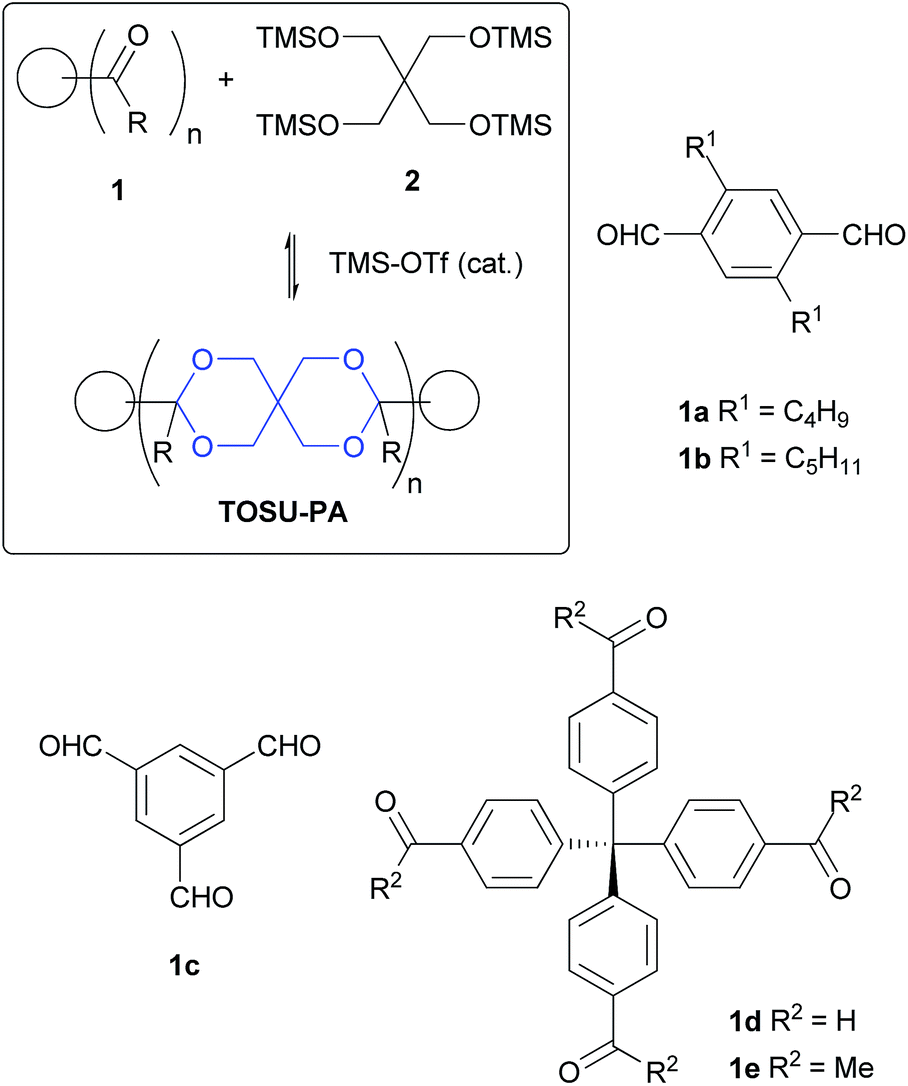 | ||
| Scheme 1 Synthesis of TOSU-polyacetals and structure of building blocks 1a–e (the TOSU skeleton is marked blue in the formula of TOSU-PA). | ||
1D polyacetals
A linear 1D model system was analyzed first to investigate the optimum conditions and the mechanism of the polyacetalization, especially the issue of reversibility. Reaction between terephthalaldehydes 1a, b, bearing solubility enhancing alkyl groups in 2- and 5-position,20 and tetrasilylether 2 under Noyori conditions gave the polymers TOSU-PA-a and -b respectively (Scheme 2). Compounds TOSU-PA-a, b are white solids, which are soluble in dichloromethane. Investigation with MALDI-TOF mass spectrometry (for details see the Experimental section) revealed the molar mass dispersion of the products, which have up to 10 monomer units. The maximum of the distribution was centred around 4–6 monomer repeat units. Exemplarily, the MALDI-TOF MS spectrum of TOSU-PA-a (R1 = C4H9) is depicted in Fig. 2. The most intensive peaks in the mass spectra (marked with ♦) originate from polymer TOSU-PA-a-A, bearing free formyl groups in the terminal positions (as Na+ adducts).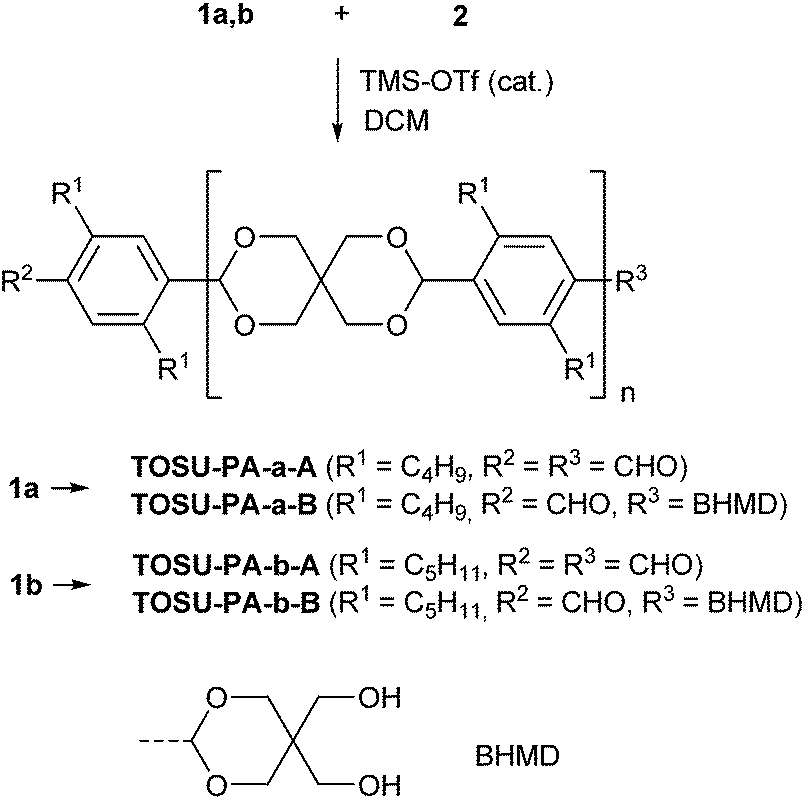 | ||
| Scheme 2 Synthesis of 1D polyketals TOSU-PA-a and TOSU-PA-b. | ||
 | ||
| Fig. 2 MALDI-TOF MS spectrum of polyacetal TOSU-PA-a (♦ = a-A, ● = a-B). | ||
 | ||
| Scheme 3 Structure of TOSU-PA-sc from the scrambling experiment. | ||
The next lower peaks (marked with ●) have a difference of 118 Dalton relative to the main peaks and can be explained by replacement of one of the formyl groups in TOSU-PA-a-A by a 5,5-bis-hydroxymethyl-1,3-dioxan-2-yl (BHMD) group (TOSU-PA-a-B). An analogous composition is observed in the case pentyl-substituted polymer TOSU-PA-b. The dependency of the degree of polymerization on the concentration of the monomers and/or the catalyst was analyzed subsequently. All synthesized polymers TOSU-PA-a, b, sc (sc = scrambling, Scheme 3) were analyzed by MALDI-TOF-MS. Minimum influence of the monomer concentration on the degree of polymerization was found (Fig. 3). Monomer concentrations ranging from 6.67 mmol L−1 up to 40 mmol L−1 of monomer 1a yielded rather similar MALDI-TOF-MS spectra. Furthermore, we varied the concentration of the catalyst TMS-OTf and observed once more no significant influence on the degree of polymerization (see ESI†). We also investigated the monomer molecular ratio. But contrary to expectations that polycondesations gave the best turnover to high molecular weights at a monomer molar ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 we determined for our soluble systems that a monomer molar ratio of 1a/b:2 = 1:1.5 gave the highest molecular weights, i.e. largest number of repeating units according to MALDI-TOF analysis.
:1 we determined for our soluble systems that a monomer molar ratio of 1a/b:2 = 1:1.5 gave the highest molecular weights, i.e. largest number of repeating units according to MALDI-TOF analysis.
 | ||
| Fig. 3 MALDI-TOF MS spectra demonstrating the influence on the monomer concentration of polyacetal TOSU-PA-a. | ||
The acetalization reaction is basically a reversible reaction. Evidence came from a scrambling experiment, in which preformed OSKs having different alkyl side chain length (TOSU-PA-a and TOSU-PA-b) were reacted with 2 and the corresponding monomer 1b or 1a under Noyori conditions. If the reaction is indeed reversible, the final product should have both butyl and pentyl substituted units as repeating units in a single chain. This was indeed found and evidenced by MALDI-TOF-MS (see Fig. 4).
 | ||
| Fig. 4 MALDI-TOF MS spectrum of the scrambling with monomer 1b and linear polymer TOSU-PA-a. | ||
Fig. 4 shows a part of the resulting MALDI-TOF-MS spectrum around repeating units with n = 4 and n = 5. The significant new peaks around the main peak of TOSU-PA-a with a mass difference of 28 Dalton are the evidence of the exchange of the given butyl side chains in TOSU-PA-a with pentyl side chains of monomer 1b.
Microporous polyacetal networks
Having established the synthetic route towards OSK, we aimed the synthesis of microporous OSK networks (TOSU-PA-c to TOSU-PA-e) by reaction of multifunctional aldehydes or ketones (1c–1e) with 2 under Noyori conditions. First of all we tried to use the reaction conditions, which have been shown suitable for the synthesis of soluble TOSU-PAs. However, it was impossible to obtain micro- or mesoporous solids using those conditions, but soluble products. Instead, we used ultrasonic irradiation based on experiences that showed the possibility of conducting polymerization reactions using ultrasonic irradiation.25 It turned out that the products obtained by using ultrasonic irradiation yielded indeed cross-linked micro- or mesoporous solid products. The choice of the solvent and the used catalyst did also seriously impact the final porosity of the polymer networks (see below). Depending on the conditions, which ultimately lead to phase separation, we observed also meso- and/or macroporosity in some of the materials. The finer effects of the phase separation and the impact of the reversibility of the reaction are however hard to discriminate in their effect on the meso/macroporosity and we will not discuss them in detail within this study. Initial experiments were conducted using 1,3,5-triformyl benzene (1c) as building block (see Scheme 4). | ||
| Scheme 4 Synthesis of TOSU-PA-c. | ||
Fig. 5 shows the N2 (77.4 K) and CO2 (273 K) adsorption/desorption isotherms of TOSU-PA-c and TOSU-PA-d materials synthesized under varying conditions.
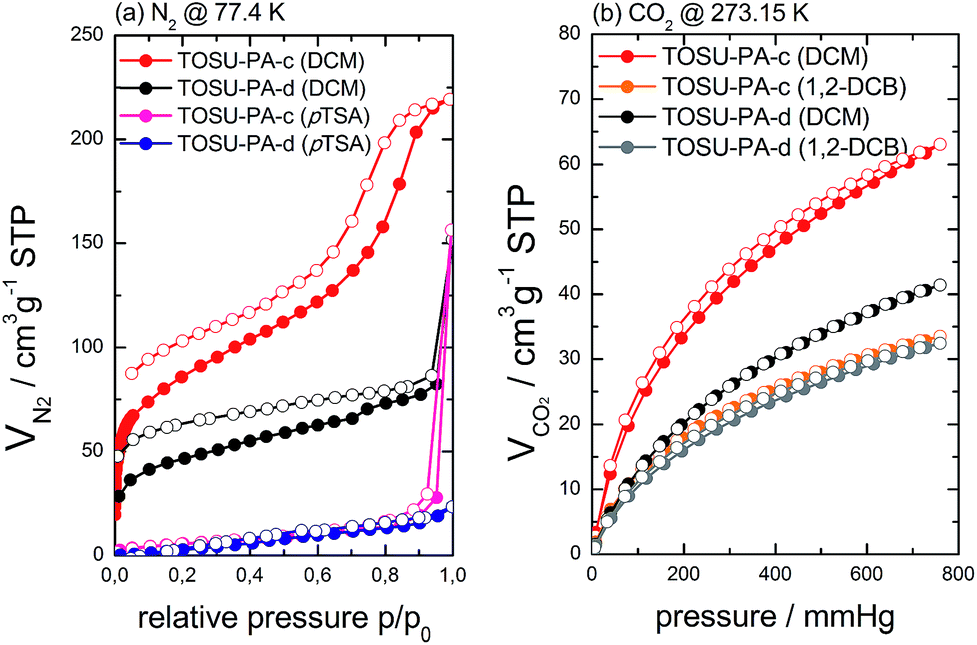 | ||
| Fig. 5 N2 (77.4 K) (a) and CO2 (273 K) (b) adsorption/desorption isotherms (adsorption with filled spheres, desorption with hollow spheres) of various TOSU-PA-c/TOSU-PA-d materials synthesized under different conditions. | ||
A first question relates to the used acetalization conditions. Fig. 5a shows clearly, that only materials synthesized under Noyori conditions (using DCM as solvent in first instance) yield porous materials. Classic conditions (i.e. refluxing benzene under continuous water removal and catalysis by 4-toluenesulfonic acid, pTSA) did not result in micro/mesoporous materials. This is in line with a report of Han et al., who could show that rather harsh conditions were needed to obtain microporous polyketals based on pTSA catalysis (180 °C, 3d, vacuum, closed flask).26
A closer look on the N2 adsorption/desorption isotherms shows that TOSU-PA-c shows some significant mesoporosity (indicated by the very steep volume increase at p/p0 ∼ 0.8 and the associated hysteresis) next to pronounced microporosity, which is evident from the large volume uptake at low relative pressures but also from more detailed isotherm analysis of this particular material (see ESI†). TOSU-PA-d does not show signs of mesoporosity. At this stage, we will not discuss the mesoporosity in detail as it seems to originate from the finer details of the phase separation, which might vary from batch to batch. Instead, we wish to discuss the microporosity in more detail, which is expected to originate from the rigid molecular structure of the used monomers and the linking chemistry (although phase separation might influence the final microporosity to some extent as well).
Associated with the microporosity of the materials, a non-closing hysteresis loop (i.e. adsorption and desorption branch do not unify even at very low relative pressure) is observed, which is a characteristic feature of many microporous polymers.27 The hysteresis was discussed in terms of the micropore connectivity and the softness of the materials and it was shown that CO2 adsorption at 273 K is another (more) useful tool for the analysis of microporous polymers. The materials were hence analysed by CO2 adsorption/desorption (see Fig. 5b for examples) as well and the impact of changed synthesis conditions was analysed as well.
In a first experiment, the CO2 adsorption capacity of TOSU-PA-c and TOSU-PA-d synthesized under Noyori conditions using either DCM or 1,2-dichlorobenzene (1,2-DCB) as solvent, was investigated (Fig. 5b). The results for DCM based systems are in line with the trend obtained by N2 physisorption at 77.4 K, i.e.TOSU-PA-c (DCM) and TOSU-PA-d (DCM) show high microporosity with the triformyl benzene based system having a higher porosity (CO2 uptake of 2.85 mmol g−1vs. 1.85 mmol g−1 at 273 K and 1 bar). The use of 1,2-dichlorobenzene (1,2-DCB) resulted finally in materials with lowered porosity. Interestingly, the porosity was quite comparable for both materials (TOSU-PA-c and TOSU-PA-d) in this case. This could relate to the fact that upon synthesis in 1,2-DCB no solid product precipitated from the reaction mixture (in contrast to the use of DCM), but just upon addition of ethanol. The materials obtained by this method could however not be dissolved after drying under high vacuum (see ESI† for details). The fact that no precipitate was formed initially indicates however a rather incomplete reaction, which in turn provides an explanation for the lowered porosity.28 It should be mentioned that 1,4-dichloro benzene (1,4-DCB) can basically also be used as solvent that allows the synthesis of microporous TOSU-PA-c. Its use under Noyori conditions is however not practically useful. This is due to its solid nature, which results in rather heterogenous conditions. 1,4-DCB was hence not investigated any further.
Finally, monomer 1e was used to produce TOSU-PA-e. 1e is basically a somewhat less reactive, but more readily available keto-based monomer. Reaction in 1,2-DCB yielded a moderately microporous polymer, while reaction in DCM led surprisingly to a lowered porosity (see ESI† for isotherm data) This might be related to the different solubility of the keto monomer 1e and oligomers build from it, compared to the formyl based monomer 1d. Table 1 summarizes the CO2 uptake for all materials as well as the respective surface areas and pore volume data obtained by analysis of the CO2 data by GCMC methodology,29 or by analysis of the N2 data by QSDFT analysis.30
| Material | S BET a/m2 g−1 | V pore a/cm3 g−1 | CO2 capacityb/mmol g−1 |
|---|---|---|---|
| a From N2 data at 77.4 K, pore volume determined from uptake at p/p0 = 0.95. b At 273.15 K and 1 bar. | |||
| TOSU-PA-1c (DCM) | 310 | 0.340 | 2.81 |
| TOSU-PA-1c (1,2-DCB) | n.d. | n.d. | 1.52 |
| TOSU-PA-1c (pTSA) | 22 | n.d. | n.d. |
| TOSU-PA-1d (DCM) | 233 | 0.128 | 1.85 |
| TOSU-PA-1d (1,2-DCB) | n.d. | n.d. | 1.47 |
| TOSU-PA-1d (pTSA) | 25 | n.d | 0.67 |
| TOSU-PA-1e (DCM) | 14 | n.d. | 0.71 |
| TOSU-PA-1e(1,2-DCB) | 38 | n.d | 1.38 |
The structures of prepared TOSU-PAs were confirmed by FT-IR spectroscopy. In all spectra of the materials, bands that can be attributed to the typical bands of the monomers e.g. carbonyl stretching of aldehyde and acetyl monomers at 1690 cm−1 and the C–H stretching at 2950 cm−1 (Fig. S6–S10, ESI†) are observed. The new C–O–C–O–C stretching vibration at 1160 cm−1 indicates a successful acetalization.
Finally, thermal analysis of selected materials indicated a reasonable stability (see ESI†). The onset of decomposition under air was found at temperatures larger than 200 °C. Decomposition took place in a number of distinct steps, which can be related to the chemical diversity of aliphatic and aromatic units, connected by heteroatoms. Full decomposition was usually observed under air atmosphere at temperatures of ∼650 °C, indicating that no inorganic residues are contained within the materials. The thermal stability under nitrogen atmosphere was tested for a single material (TOSU-PA-e) as well. The onset of decomposition was again found at T > 250 °C, which is not very much different compared to decomposition under air, indicating that the initial steps are thermolytic cleavage of covalent bonds. Again, multiple further decomposition steps were found, however the material cannot be decomposed completely under N2 atmosphere and 32% residue (most probably carbon materials) are found at T ∼ 1000 °C.
It should be noted that the obtained TOSU-PAs are totally amorphous, concluded from Wide-Angle X-ray Scattering (WAXS) measurements (see ESI†).
Conclusions
In summary, we have shown that the acetalization of various carbonyl compounds, each with 2–4 carbonyl groups, with tetrasilylether 2 provides polyacetals (PA) under very mild conditions (short reaction time, room temperature). The central structural element of these molecules is the conformationally rigid 2,4,8,10-tetraoxaspiro[5.5]undecane (TOSU) skeleton. In the case of tri- and tetravalent carbonyl compounds (1c–e) we obtained TOSU-PAs with moderately high porosity. They show however also a sensitivity of the observed porosity to the exact reaction conditions and it can be speculated that this complexity arises as a consequence of the different solubility of the growing branched/cross-linked polymer in the various solvents, which impairs the phase-separation process as well as the principal reversibility of the reaction. Given the high CO2 capacity of some of the materials, we believe that the presented platform of smooth and simple acetalization reactions is indeed of interest for further development of porous materials.Experimental section
Materials and methods
All chemicals were purchased from Aldrich or Merck and were used as received. 2,5-Dibutylbenzene-1,4-dicarbaldehyde (1a) and 2,5-dipentylbenzene-1,4-dicarbaldehyde (1b) were synthesized according to the reported procedure, respectively.20 Benzene-1,3,5-tricarbaldehyd 1c also was synthesized to a reported procedure.31 And tetrakis(4-acetylphenyl)methane (1e) was synthesized to the reported procedure of Müller et al.32 The detailed synthesis procedure of 4,4′,4′′,4′′′-methane-tetrayltetrabenzaldehyde (1d) is included in the supporting information.Preparation of TOSU-PA-a
To a solution of 123 mg of 1a (0.5 μmol, 1.0 eq.) and 318 mg of 2 (0.75 μmol, 1.5 eq.) in 15 mL dry dichloromethane were added 100 μL TMSOTf. The resulting mixture was stirred for 20 h at 20 °C. The solution was neutralized with a saturated solution of sodium bicarbonate, dried with magnesium sulphate and evaporated. The crude product was recrystallized from ethanol and was obtained after filtration as white solid.The same procedure is followed for the preparation of TOSU-PA-b.
Preparation of TOSU-PA-sc
To a solution of 24 mg of TOSU-PA-a, 6.5 mg of 1b (8.9 μmol) and 3.8 mg of 2 (8.9 μmol) in 5 mL dry dichloromethane were added 20 μL TMSOTf. The resulting mixture was stirred for 20 h at 20 °C. The solution was neutralized with a saturated solution of sodium bicarbonate, dried with magnesium sulphate and the solvent was evaporated off. The crude product was recrystallized from ethanol and was obtained after filtration as white solid.Preparation of TOSU-PA-c
To a solution of 100 mg of 1c (617 μmol, 1 eq.) and 393 mg of 2 (925 μmol, 1.5 eq.) in 50 mL dry dichloromethane were added 100 μL TMSOTf. After the reaction in an ultrasonic bath for 20 h the obtained white-gray precipitate was filtered and washed with sodium bicarbonate, water, ethyl acetate, acetone, dichloromethane and dried in high vacuum.The same reaction conditions in 1,2-DCB as solvent did not lead to a solid, but with the addition of ethanol after reaction there could be obtained a gray precipitate which was filtered and washed with sodium bicarbonate, water, ethyl acetate, acetone, dichloromethane and dried in high vacuum.
The same procedure is followed for the preparation of TOSU-PA-d–e.
Instrumentation
Nitrogen (N2) adsorption/desorption experiments were performed with a Quantachrome Quadrasorb machine at liquid nitrogen temperature (77.4 K), CO2 adsorption was measured at 273.15 K using a Quantachrome Autosorb-MP1 machine. High-purity gases were used in all cases. All samples were degassed at 70 °C for 20 hours before gas adsorption measurements. Initial data analysis (calculation of surface area using the Brunauer–Emmett–Teller (BET) equation, pore volume calculation and analysis using GCMC or QSDFT methodologies) was performed using the Quadrasorb 5.05 software package (Quantachrome Instruments). MALDI-TOF spectra were measured with the MALDI-TOF massspectrometer Axima Assurance from Shimadzu Biotech and were plotted with the software Launchpad 2.Fourier-Transform Infrared (FTIR) spectroscopy was conducted using a Perkin Elmer UATR Two machine with single reflection diamond. Wide-Angle X-ray Scattering (WAXS) was measured using a FR590 diffractometer from ENRAF NONIUS (Cu-K-alpha).
Thermogravimetric analysis (TGA) was performed under synthetic air or nitrogen flow using a NETZSCH TG209-F1. A heating rate of 10 K min−1 was used.
Notes and references
- B. H. Davis and K. S. W. Sing, Historical Aspects, in Handbook of Porous Solids, ed. F. Schüth, K. S. W. Sing and J. Weitkamp, Wiley-VCH Verlag GmbH, Weinheim, Germany, 2002 Search PubMed.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334–2375 CrossRef CAS PubMed.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705–714 CrossRef CAS PubMed.
- K. Sumida, D. L. Rogow, J. A. Mason, T. M. McDonald, E. D. Bloch, Z. R. Herm, T.-H. Bae and J. R. Long, Chem. Rev., 2012, 112, 724–781 CrossRef CAS PubMed.
- V. Davankov and M. P. Tsvurupa, Hypercrosslinked Polymeric Networks and Adsorbing Materials: Synthesis, Properties, Structure, and Applications, Elsevier, 2010 Search PubMed.
- A. Thomas, Angew. Chem., Int. Ed., 2010, 49, 8328–8344 CrossRef CAS PubMed.
- A. Thomas, P. Kuhn, J. Weber, M.-M. Titirici and M. Antonietti, Macromol. Rapid Commun., 2009, 30, 221–236 CrossRef CAS PubMed.
- N. B. McKeown and P. M. Budd, Macromolecules, 2010, 43, 5163–5176 CrossRef CAS.
- Y. Xu, M. D. Smith, M. F. Geer, P. J. Pellechia, J. C. Brown, A. C. Wibowo and L. S. Shimizu, J. Am. Chem. Soc., 2010, 132, 5334–5335 CrossRef CAS PubMed.
- T.-J. Hsu, F. W. Fowler and J. W. Lauher, J. Am. Chem. Soc., 2012, 134, 142–145 CrossRef CAS PubMed.
- R. Dawson, A. I. Cooper and D. J. Adams, Prog. Polym. Sci., 2012, 37, 530–563 CrossRef CAS PubMed.
- S. Xu, Y. Luo and B. Tan, Macromol. Rapid Commun., 2013, 34, 471–484 CrossRef CAS PubMed.
- T. Tozawa, J. T. A. Jones, S. I. Swamy, S. Jiang, D. J. Adams, S. Shakespeare, R. Clowes, D. Bradshaw, T. Hasell, S. Y. Chon, C. Tang, S. Thompson, J. Parker, A. Trewin, J. Basca, A. M. Z. Slawin, A. Steiner and A. I. Cooper, Nat. Mater., 2009, 8, 973–978 CrossRef CAS PubMed.
- M. Mastalerz and I. M. Oppel, Angew. Chem., Int. Ed., 2012, 51, 5252–5255 CrossRef CAS PubMed.
- Reviews on molecular rods: (a) J. M. Tour, Chem. Rev., 1996, 96, 537–553 CrossRef CAS PubMed; (b) P. F. H. Schwab, M. D. Levin and J. Michl, Chem. Rev., 1999, 99, 1863–1933 CrossRef CAS PubMed; (c) M. D. Levin, P. Kaszynski and L. Michl, Chem. Rev., 2000, 100, 169–234 CrossRef CAS PubMed; (d) J. M. Tour, Acc. Chem. Res., 2000, 33, 791–804 CrossRef CAS PubMed; (e) P. F. H. Schwab, J. R. Smith and J. Michl, Chem Rev., 2005, 105, 1197–1279 CrossRef CAS PubMed; (f) N. Sakai, J. Mareda and S. Matile, Acc. Chem. Res., 2005, 38, 79–87 CrossRef CAS PubMed.
- P. Wessig, K. Möllnitz and C. Eiserbeck, Chem.–Eur. J., 2007, 13, 4859–4872 CrossRef CAS PubMed.
- P. Wessig and K. Möllnitz, J. Org. Chem., 2008, 73, 4452–4457 CrossRef CAS PubMed.
- P. Müller, J. Nikolaus, S. Schiller, A. Herrmann, K. Möllnitz, S. Czapla and P. Wessig, Angew. Chem., Int. Ed., 2009, 48, 4433–4435 CrossRef PubMed.
- J. Nikolaus, S. Czapla, K. Möllnitz, C. T. Höfer, A. Herrmann, P. Wessig and P. Müller, Biochim. Biophys. Acta, Biomembr., 2011, 2781–2788 CrossRef CAS PubMed.
- P. Wessig and K. Möllnitz, J. Org. Chem., 2012, 77, 3907–3920 CrossRef CAS PubMed.
- A. Techen, S. Czapla, K. Möllnitz, D. Budach, P. Wessig and M. Kumke, Helv. Chim. Acta, 2013, 96, 2046–2067 CrossRef CAS.
- C. Grimm, T. Meyer, S. Czapla, J. Nikolaus, H. A. Scheidt, A. Vogel, A. Herrmann, P. Wessig, D. Huster and P. Müller, Chem.–Eur. J., 2013, 19, 2703–2710 CrossRef CAS PubMed.
- T. Tsunoda, M. Suzuki and R. Noyori, Tetrahedron Lett., 1980, 21, 1357–1358 CrossRef CAS.
- S. Aldridge, R. J. Calder, A. Rossin, A. A. Dickinson, D. J. Willock, C. Jones, D. J. Evans, J. W. Steed, M. E. Light, S. J. Coles and M. B. Hursthouse, J. Chem. Soc., Dalton Trans., 2002, 2020–2026 RSC.
- P. Kmus and T. J. Patraboy, J. Phys. Chem., 1985, 89, 3379–3384 CrossRef.
- Y. Han, L.-M. Zhang, Y.-C. Zhao, T. Wang and B.-H. Han, ACS Appl. Mater. Interfaces, 2013, 5, 4166–4172 CAS.
- J. Jeromenok and J. Weber, Langmuir, 2013, 29, 12982–12989 CrossRef CAS PubMed.
- A high degree of conversion is usually helpful for high surface area materials, see e.g. J. Weber and Q. B. Meng, Encycl. Polym. Sci. Technol., 2014 DOI:10.1002/0471440264.pst622 , in press.
- A. Vishnyakov, P. Ravikovitch and A. Neimark, Langmuir, 1999, 15, 8736–8742 CrossRef CAS.
- J. Landers, G. Y. Gor and A. Neimark, Colloids Surf., A, 2013, 437, 3–32 CrossRef CAS PubMed.
- M. Fourmigué, I. Johannsen, K. Boubekeur, C. Nelson and P. Batail, J. Am. Chem. Soc., 1993, 115, 3752–3759 CrossRef.
- T. J. Zimmermann, O. Freundel, R. Gompper and T. J. J. Müller, Eur. J. Org. Chem., 2000, 19, 3305–3312 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional synthetic procedures and analytical data (IR, TGA, gas adsorption analysis). See DOI: 10.1039/c4ra04437a |
| ‡ Present address: Hochschule Zittau/Görlitz, Fachgruppe Chemie, Theodor-Körner-Allee 16, D-02763 Zittau, Germany. E-mail: j.weber@hszg.de; Fax: +49-3583-61-1740; Tel: +49-3583-61-1705. |
| This journal is © The Royal Society of Chemistry 2014 |