Metal-free synthesis of methylene-bridged bis-1,3-dicarbonyl compounds via oxidative C–C bond cleavage of tertiary aliphatic amines†
Li-Juan Xing,
Xi-Mei Wang,
Hong-Ying Li,
Wen Zhou,
Ning Kang,
Peng Wang and
Bin Wang*
State Key Laboratory of Medicinal Chemical Biology and College of Pharmacy, Nankai University, Tianjin, 300071, China. E-mail: wangbin@nankai.edu.cn; Fax: +86-22-2350-7760; Tel: +86-22-2350-6290
First published on 9th June 2014
Abstract
A metal-free Bu4NI mediated oxidative reaction utilizing tertiary aliphatic amines and 1,3-dicarbonyl compounds for the synthesis of methylene-bridged bis-1,3-dicarbonyl compounds has been developed. This reaction involved an unexpected C–C bond cleavage of tertiary aliphatic amines. With this approach, the completely regioselective functionalization of β-carbon on tertiary amines was realized.
Oxidative functionalization of α-carbon in tertiary amines with subsequent formation of C–C or C–heteroatom bonds is an attractive transformation in organic synthesis.1 This approach, termed cross-dehydrogenative coupling (CDC) reaction, allows use of less functionalized reagents and often reduces the number of steps to the target molecule.2 In most cases, N,N-dialkylanilines or tetrahydroisoquinolines are used as coupling partners (Fig. 1a), which have proved to be very efficient for the generation of electrophilic iminium ions in the presence of metal catalysts and terminal oxidants.1,2 These highly reactive iminium ion intermediates can be readily trapped by a variety of nucleophiles (step a). In some cases, the coupling product A was subsequently converted to a methylene-bridged compound B through a sequential CDC reaction and C–N bond cleavage (step b).3
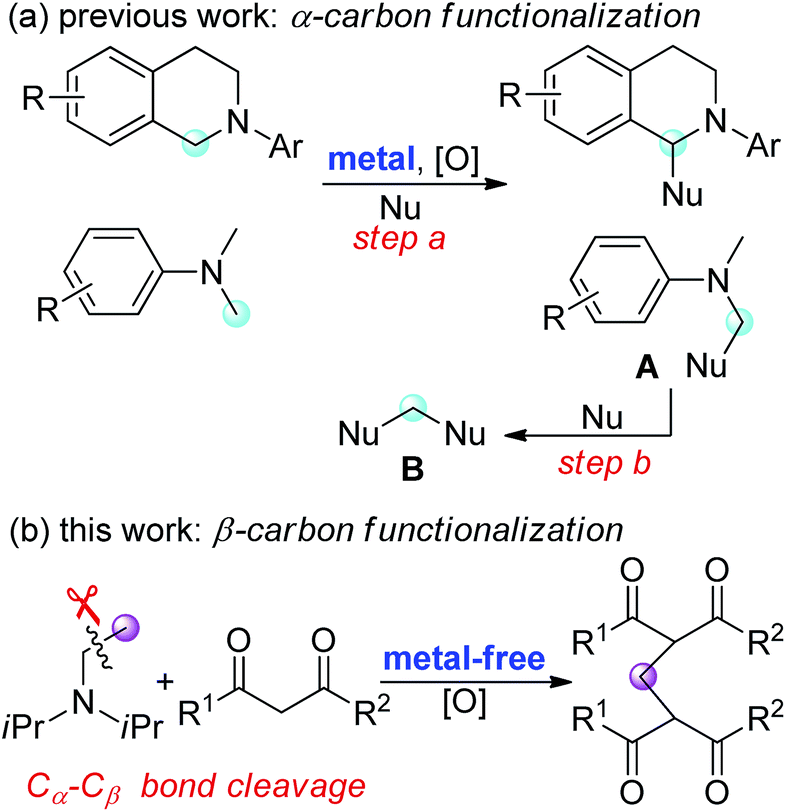 | ||
| Fig. 1 Oxidative functionalization of tertiary amines. | ||
While a variety of synthetic methods are available for oxidative coupling of N-methyl tertiary aromatic amines and the reactions occur regioselectively at α-carbon (Fig. 1a), there are relatively few general and practical approaches for the oxidative functionalization of tertiary aliphatic amines, particularly the selective functionalization of β-carbon in non-N-methyl-substituted trialkylamines.4 Moreover, the current oxidative coupling reactions of tertiary amine require metal catalysts, regardless of whether the reaction occurs at α-carbon or β-carbon. The development of metal-free oxidative coupling reactions with complete regioselectivity and environmental benign oxidants would be highly desirable.
In continuation of our work on the development of green and efficient synthetic methods for the oxidative coupling reactions,5 we have recently reported quaternary ammonium iodides mediated C3-selective formylation of indoles using N-methyl anilines as the carbonyl source.5b,c In this study, we demonstrate a Bu4NI-catalyzed oxidative coupling of tertiary aliphatic amines with 1,3-dicarbonyl compounds. This metal-free transformation, involving unprecedented C–C bond cleavage of trialkylamines, provides an efficient access to methylene-bridged bis-1,3-dicarbonyl compounds, which are valuable precursors for heterocycles and materials synthesis3c,6 (Fig. 1b).
Our initial studies focused on Bu4NI-catalyzed oxidative-coupling of 1,3-diphenyl-1,3-propanedione 1a with a variety of different tertiary amines in the presence of aqueous tert-butyl hydroperoxide (TBHP).7 All reactions was carried out in 1,4-dioxane at 25 °C for 24 h or 60 °C for 5 h, and the results were shown in Table 1. The oxidative coupling reaction of 1a with N,N-dimethylaniline 2a as the carbon source afforded methylene-bridged product 3a in 25% yield at 25 °C, while higher temperature led to complex reaction mixtures and compound 3a was not detected (Table 1, entry 1). The reaction produced 3a in a moderate yield (48%) at 25 °C when 4-methyl-N,N-dimethylaniline 2b was used as the amine component, but it also proved to be unsuccessful at 60 °C (Table 1, entry 2). Both N,N-diethylaniline 2c and N,N-dimethylbenzylamine 2d were ineffective for this transformation at 25 °C and 60 °C (Table 1, entries 3 and 4). We then investigated tertiary aliphatic amines in this oxidative system. To our delight, the reaction of 1a with triethylamine 2e generated the product 3a in 38% and 16% yields, respectively (Table 1, entry 5). It is noted that ethyl bis-adduct 4e, described by Li and coworkers in their iron-catalyzed oxidative reactions (Fig. 2),8 was not detected in the reaction mixture. Further investigation revealed that N,N-diisopropylethylamine 2f (DIPEA) reacted smoothly with substrate 1a to give the product 3a in good yields under different conditions (Table 1, entry 6). In sharp contrast, when N,N-diisopropylmethylamine 2g or N,N-diethylmethylamine 2h was subjected to the present oxidative conditions, the corresponding product 3a was not detected at all, regardless of changes in the reaction temperature or time (Table 1, entries 7 and 8). Very interestingly, tertiary amines with long alkyl chain such as tripropylamine 2i and tributylamine 2j could also afford the methylene-bridged product 3a, albeit in very low yields (Table 1, entries 9 and 10). In light of the success with DIPEA, we envisioned that bulky amines could be beneficial to the reaction. Two sterically hindered tertiary amines 2k and 2l were then investigated respectively. In the case of N-cyclohexyl-N-methylcyclohexanamine 2k, the desirable product 3a was formed only when the reaction was performed at 25 °C (Table 1, entry 11). In contrast, another N-ethyl substituted amine 2l afforded 3a in moderated yields under different conditions (Table 1, entry 12). It is noted that secondary amines are ineffective for the formation of 3a.9
|
|||
|---|---|---|---|
| Entry | Amine | Yieldb | Yieldc |
| a Reaction conditions: 1,3-diketone 1a (0.5 mmol), amine(1.5 mmol), tert-butyl hydrogenperoxide (TBHP, 2.0 mmol, 70% aqueous solution), tetrabutylammonium iodide (Bu4NI, 0.1 mmol, 36.9 mg) in 1,4-dioxane (1 mL).b Isolated yield based on compound 1a at 25 °C, 24 h.c Isolated yield based on compound 1a at 60 °C, 5 h. | |||
| 1 | PhNMe2 2a | 25% | 0 |
| 2 | TolNMe2 2b | 48% | 0 |
| 3 | PhNEt2 2c | 0 | 0 |
| 4 | BnNMe2 2d | 0 | 0 |
| 5 | Et3N 2e | 38% | 16% |
| 6 | (i-Pr)2NEt 2f | 72% | 73% |
| 7 | (i-Pr)2NMe 2g | 0 | 0 |
| 8 | Et2NMe 2h | 0 | 0 |
| 9 | (n-Pr)3N 2i | 3% | 9% |
| 10 | (n-Bu)3N 2j | Trace | 10% |
| 11 | Cy2NMe 2k | 26% | 0 |
| 12 | Cy2NEt 2l | 38% | 59% |
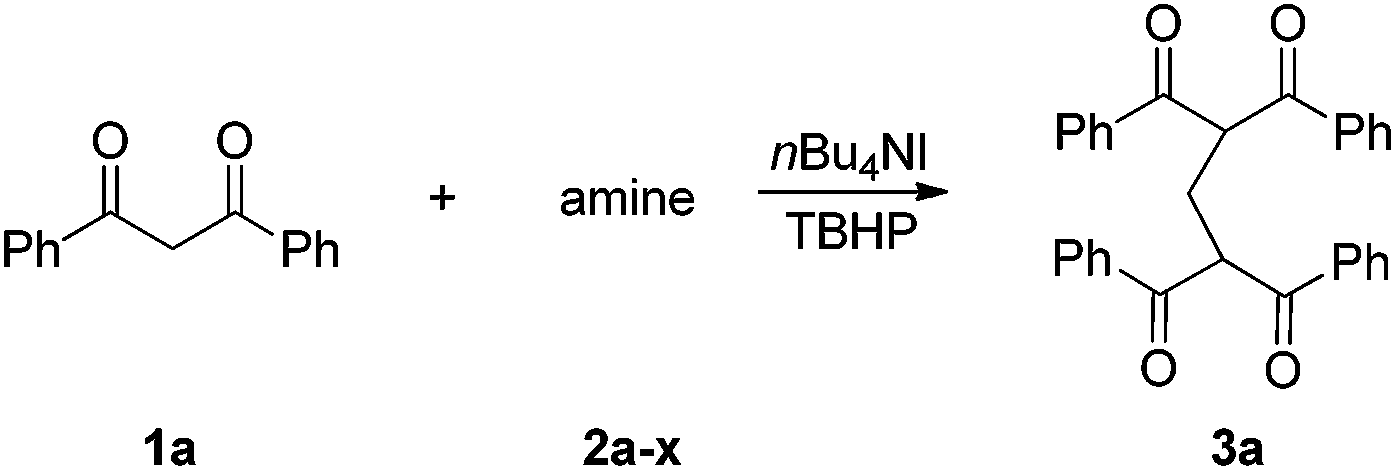
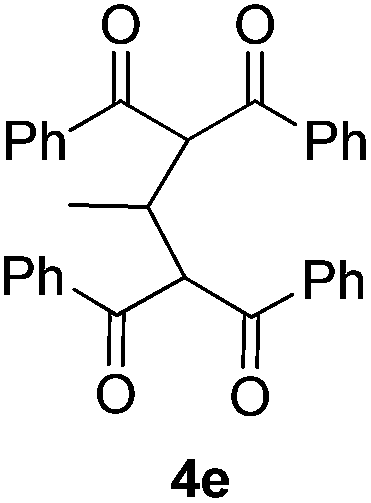 | ||
| Fig. 2 Possible ethyl bis-adduct 4e. | ||
| a Reaction conditions: 1,3-dicarbonyl compound 1 (0.5 mmol), amine (1.5 mmol), TBHP (2.0 mmol, 70% aqueous solution), Bu4NI (0.1 mmol, 36.9 mg) in 1,4-dioxane (1 mL), 60 °C, 5 h, isolated yield based on compound 1. |
|---|
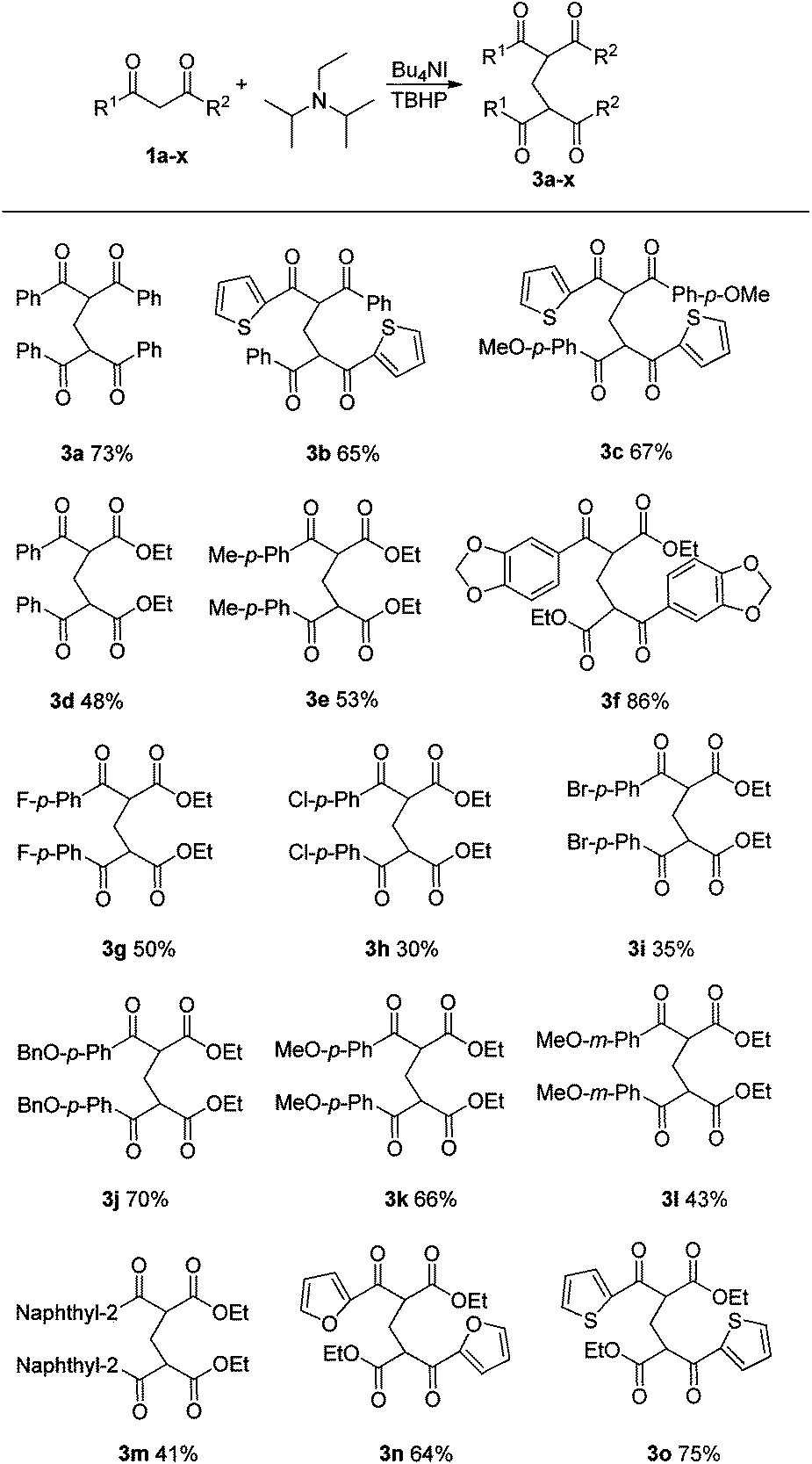 |
On the basis of the results achieved with DIPEA, a range of 1,3-dicarbonyl compounds 1 was next examined under optimal conditions. In addition to 1,3-diphenyl-1,3-propanedione 1a, dissymmetrical 1,3-diketones such as 1b and 1c were also successfully applied in this transformation (Table 2, 3b and 3c). The present oxidative reaction catalyzed by Bu4NI was easily extended to β-ketoesters substrates (Table 2, 3d–o). Aromatic β-ketoesters bearing electron-donating (3e–f, 3j–l) or electron-withdrawing groups (3g–i) reacted with DIPEA smoothly, affording the corresponding products in 30–86% yields. However, substrates bearing electron-withdrawing groups gave lower yields.
 | (1) |
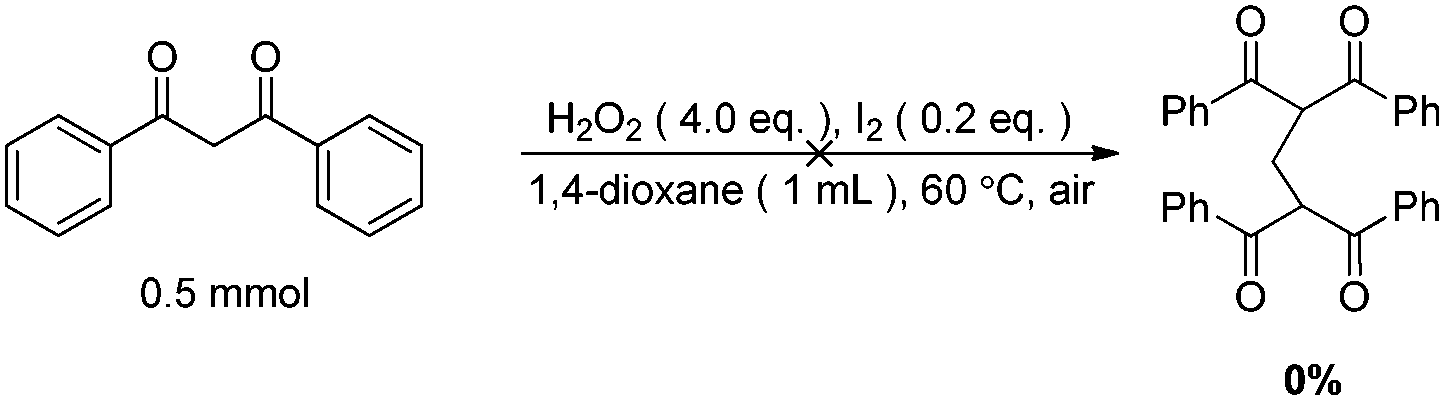 | (2) |
 | (3) |
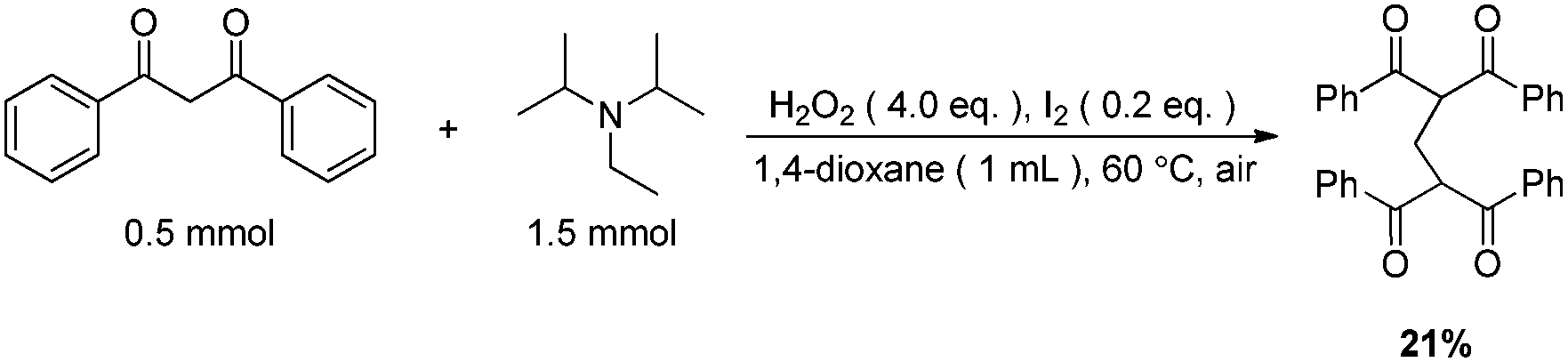
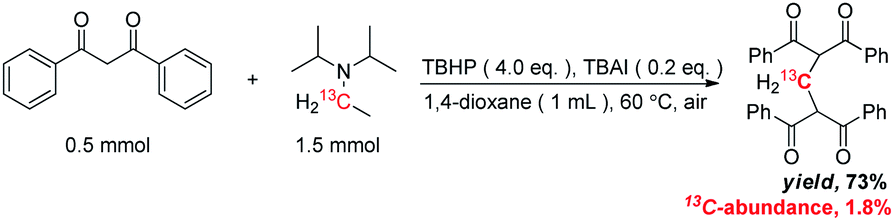
The origin of methylene carbon in product 3a was the major concern in our mechanistic study. Treatment of 1a with H2O2/I2 gave 3a in 21% yield in the presence of DIPEA (eqn (1)). In contrast, a similar reaction without DIPEA did not take place at all (eqn (2)). Obviously, DIPEA was the methylene donor in the reaction, and the possibility of generating methylene carbon from TBHP and Bu4NI could be excluded. To further investigate the information of methylene group in 3a, 1–13C-labeled DIPEA was subjected to the reaction conditions. To our surprise, only 1.8% incorporation at methylene carbon was observed (eqn (3)). The results unambiguously established that the carbon atom of methylene group was originated from β-carbon of DIPEA, rather than α-carbon. Clearly, the Cα–Cβ bond of DIPEA was cleaved, resulting in the formation of methylene-bridged bis-1,3-dicarbonyl compound 3. Further mechanistic investigation indicated that the formation of product 3a was suppressed in the presence of radical scavenger 2,2,6,6-tetramethyl-1-piperidinyloxy (TEMPO). To our knowledge, alkyl hypoiodite can be generated from alkyl iodide by oxidation and rearrangement.10 The homolytic cleavage of O–I bond can provide an alkoxyl free radical.11 This radical could undergo β-scission to give a carbonyl compound and another alkyl iodide.12 In the present reaction, amides were detected through 13C NMR experiments during the course of the reaction (Fig. 1S†). On the basis of earlier studies and results obtained above, we propose a plausible reaction mechanism (Scheme 1). First, DIPEA reacts with the oxidant via two single electron transfers (SET) to give iminium ion A,7b which can isomerize to enamine B. At the same time, TBHP efficiently oxidizes iodide (I−) to hypoiodous acid or molecular iodine.7b,13 Then, the reaction of iodine species with enamine B yields iodonium ion C,14 which is subsequently opened at the sterically less hindered carbon by compound 1a to give compound D. Oxidation of alkyl iodide D with TBHP affords iodoso-compound E, which readily rearranges to the hypoiodite F.10 The homolytic cleavage of O–I bond takes place to afford the alkoxy radical G,11 followed by rearrangement through C–C bond scission to provide amide H and alkyl iodide I.12 Finally, the resulting alkyl iodide I reacts with 1a by either a nucleophilic substitution or a tandem elimination/addition to afford the product 3a. In addition, it cannot be excluded that the reaction proceeds via a 1,2-dioxetane intermediate, which can be generated from enamine and oxidant.15 The active 1,2-dioxetane then undergoes C–C bond cleavage to produce formaldehyde and amide. The Knoevenagel condensation of formaldehyde with 1a gives an α,β-unsaturated dicarbonyl compound, which subsequently undergoes Michael addition with remaining excess 1a to produce the final bis adduct 3a. However, due to the change in color in the reaction, the Nash test16 is not applicable for detecting formaldehyde.
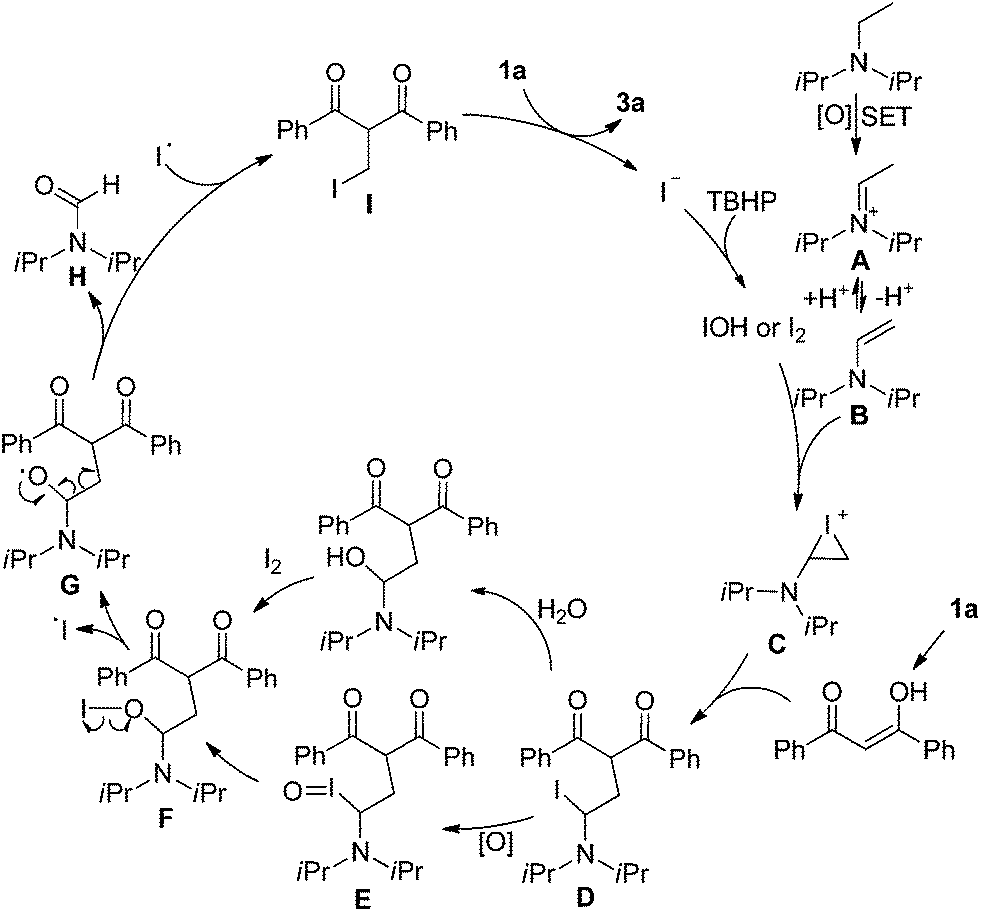 | ||
| Scheme 1 Proposed reaction mechanism. | ||
Conclusions
In conclusion, we have developed a metal-free oxidative coupling reaction mediated by Bu4NI that allows for the synthesis of methylene-bridged bis-1,3-dicarbonyl compounds from tertiary aliphatic amines and 1,3-dicarbonyl compounds. The β-carbon of DIPEA was converted to the methylene carbon through an oxidative C–C bond cleavage. We believe this C–C bond cleavage of tertiary aliphatic amine holds promise to develop other novel transformations. The methodology is currently under active investigation to explore its detailed mechanism and is also extended to the synthesis of other methylene-bridged bis adducts.Acknowledgements
Financial support by the National Science Foundation of China (no. 21172120, 20902050) and National University Student Innovation Program (BX111359, 20130055020). We thank Prof. Renhua Fan in Fudan University for helpful discussions.Notes and references
- (a) K. R. Campos, Chem. Soc. Rev., 2007, 36, 1069–1084 RSC; (b) M. Klussmann and D. Sureshkumar, Synthesis, 2011, 353–369 CrossRef CAS PubMed.
- (a) C.-J. Li, Acc. Chem. Res., 2009, 42, 335–344 CrossRef CAS PubMed; (b) C. Liu, H. Zhang, W. Shi and A. Lei, Chem. Rev., 2011, 111, 1780–1824 CrossRef CAS PubMed; (c) C. S. Yeung and V. M. Dong, Chem. Rev., 2011, 111, 1215–1292 CrossRef CAS PubMed.
- (a) H. Li, Z. He, X. Guo, W. Li, X. Zhao and Z. Li, Org. Lett., 2009, 11, 4176–4179 CrossRef CAS PubMed; (b) W.-J. Yoo, A. Tanoue and S. Kobayashi, Chem.–Asian J., 2012, 7, 2764–2767 CrossRef CAS PubMed; (c) L. Zhang, C. Peng, D. Zhao, Y. Wang, H.-J. Fu, Q. Shen and J.-X. Li, Chem. Commun., 2012, 48, 5928–5930 RSC.
- (a) X.-F. Xia, X.-Z. Shu, K.-G. Ji, Y.-F. Yang, A. Shaukat, X.-Y. Liu and Y.-M. Liang, J. Org. Chem., 2010, 75, 2893–2902 CrossRef CAS PubMed; (b) B. Sundararaju, M. Achard, G. V. M. Sharma and C. Bruneau, J. Am. Chem. Soc., 2011, 133, 10340–10343 CrossRef CAS PubMed; (c) K. Yuan, F. Jiang, Z. Sahli, M. Achard, T. Roisnel and C. Bruneau, Angew. Chem., Int. Ed., 2012, 51, 8876–8880 CrossRef CAS PubMed; (d) N. Takasu, K. Oisaki and M. Kanai, Org. Lett., 2013, 15, 1918–1921 CrossRef CAS PubMed.
- (a) J. Huang, L.-T. Li, H.-Y. Li, E. Husan, P. Wang and B. Wang, Chem. Commun., 2012, 48, 10204–10206 RSC; (b) L.-T. Li, J. Huang, H.-Y. Li, L.-J. Wen, P. Wang and B. Wang, Chem. Commun., 2012, 48, 5187–5189 RSC; (c) L.-T. Li, H.-Y. Li, L.-J. Xing, L.-J. Wen, P. Wang and B. Wang, Org. Biomol. Chem., 2012, 10, 9519–9522 RSC.
- (a) J. Guin, R. Fröhlich and A. Studer, Angew. Chem., Int. Ed., 2008, 47, 779–782 CrossRef CAS PubMed; (b) A. Cuadro, J. Elguero and P. Navarro, Chem. Pharm. Bull., 1985, 33, 2535–2540 CrossRef CAS; (c) N. M. M. Moura, M. A. F. Faustino, M. G. P. M. S. Neves, F. A. A. Paz, A. M. S. Silva, A. C. Tome and J. A. S. Cavaleiro, Chem. Commun., 2012, 48, 6142–6144 RSC; (d) A. Gogoll, C. Johansson, A. Axén and H. Grennberg, Chem.–Eur. J., 2001, 7, 396–403 CrossRef CAS; (e) D. Mousset, I. Gillaizeau, A. Sabatié, P. Bouyssou and G. Coudert, J. Org. Chem., 2006, 71, 5993–5999 CrossRef CAS PubMed; (f) D. F. Martin, M. Shamma and W. C. Fernelius, J. Am. Chem. Soc., 1958, 80, 5851–5856 CrossRef CAS; (g) R. Karvembu and K. Natarajan, Polyhedron, 2002, 21, 1721–1727 CrossRef CAS.
- (a) M. Uyanik and K. Ishihara, ChemCatChem, 2012, 4, 177–185 CrossRef CAS; (b) P. Finkbeiner and B. J. Nachtsheim, Synthesis, 2013, 45, 979–999 CrossRef CAS PubMed.
- W. Liu, J. Liu, D. Ogawa, Y. Nishihara, X. Guo and Z. Li, Org. Lett., 2011, 13, 6272–6275 CrossRef CAS PubMed.
- X. B. Zhang, M. Wang, Y. C. Zhang and L. Wang, RSC Adv., 2013, 3, 1311–1316 RSC.
- (a) H. J. Reich and S. L. Peake, J. Am. Chem. Soc., 1978, 100, 4888–4889 CrossRef CAS; (b) R. C. Cambie, D. Chambers, B. G. Lindsay, P. S. Rutledge and P. D. Woodgate, J. Chem. Soc., Perkin Trans. 1, 1980, 822–827 RSC; (c) T. L. Macdonald, N. Narasimhan and L. T. Burka, J. Am. Chem. Soc., 1980, 102, 7760–7765 CrossRef CAS.
- J. Janjatovic and Z. Majerski, J. Org. Chem., 1980, 45, 4892–4898 CrossRef CAS.
- (a) P. Gray and A. Williams, Chem. Rev., 1959, 59, 239–328 CrossRef CAS; (b) C. Walling and A. Padwa, J. Am. Chem. Soc., 1963, 85, 1593–1597 CrossRef CAS; (c) D. E. O'Dell, J. T. Loper and T. L. Macdonald, J. Org. Chem., 1988, 53, 5225–5229 CrossRef; (d) C. S. Aureliano Antunes, M. Bietti, O. Lanzalunga and M. Salamone, J. Org. Chem., 2004, 69, 5281–5289 CrossRef PubMed.
- I. Matsuzaki, T. Nakajima and H. A. Liebhafsky, Chem. Lett., 1974, 3, 1463–1466 CrossRef.
- (a) A. Yoshimura, K. R. Middleton, C. Zhu, V. N. Nemykin and V. V. Zhdankin, Angew. Chem., Int. Ed., 2012, 51, 8059–8062 CrossRef CAS PubMed; (b) J.-S. Tian, K. W. J. Ng, J.-R. Wong and T.-P. Loh, Angew. Chem., Int. Ed., 2012, 51, 9105–9109 CrossRef CAS PubMed.
- (a) N. A. Milas and B. Plesnicar, J. Am. Chem. Soc., 1968, 90, 4450–4453 CrossRef CAS; (b) W. Ando, T. Saiki and T. Migita, J. Am. Chem. Soc., 1975, 97, 5028–5029 CrossRef CAS; (c) H. H. Wasserman and J. L. Ives, J. Am. Chem. Soc., 1976, 98, 7868–7869 CrossRef CAS; (d) J. E. Huber, Tetrahedron Lett., 1968, 9, 3271–3272 CrossRef; (e) R. Lin, F. Chen and N. Jiao, Org. Lett., 2012, 14, 4158–4161 CrossRef CAS PubMed; (f) J. Basran, I. Efimov, N. Chauhan, S. J. Thackray, J. L. Krupa, G. Eaton, G. A. Griffith, C. G. Mowat, S. Handa and E. L. Raven, J. Am. Chem. Soc., 2011, 133, 16251–16257 CrossRef CAS PubMed.
- T. Nash, Biochem. J., 1953, 55, 416–421 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4ra04419c |
| This journal is © The Royal Society of Chemistry 2014 |