Dispersion-controlled PtCu clusters synthesized with citric acid using galvanic displacement with high electrocatalytic activity toward methanol oxidation†
Qing Lvab,
Jinfa Changab,
Wei Xing*a and
Changpeng Liu*c
aState Key Laboratory of Electroanalytical Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun, Jilin, China. E-mail: xingwei@ciac.jl.cn; Fax: +86-431-85262225; Tel: +86-431-85262223
bUniversity of Chinese Academy of Sciences, Beijing, China
cLaboratory of Advanced Power Sources, Changchun Institute of Applied Chemistry, 5625 Renmin Street, Changchun, Jilin, China. E-mail: liuchp@ciac.jl.cn
First published on 26th June 2014
Abstract
Dispersion-controlled carbon supported PtCu clusters were firstly synthesized using galvanic displacement of Cu/C, in which citric acid worked as the dispersion agent and its concentration was adjusted to form the as-synthesized clusters. It was found that dispersion played a significant role in tuning the activity for methanol electrooxidation.
Direct methanol fuel cells (DMFCs) hold great potential as clean energy sources for powering portable electronic devices and vehicles.1–5 Currently, Pt-based nanomaterials are the most effective electrocatalysts to facilitate the anodic methanol oxidation reaction,1,6,7 due to their unique catalytic properties in fuel cells.7–15 However, the high cost of the noble metal platinum and low activities of the anode catalysts at room temperature are still some of the important issues that hinder the commercialization of DMFCs.2,16–18 To increase the methanol oxidation activity and reduce platinum loading, bimetallic catalysts of platinum alloyed with a less expensive metal and unique morphologies are often used.1,6,7,19
It is generally known that the size, shape, composition and structure of Pt-based catalysts are important parameters that determine the catalytic activity.13,15,20–24 Galvanic replacement is an effective method for controlling the synthesis of Pt-based nanomaterials with special shapes and composition.25–27 For example, a Pt hollow nanosphere catalyst was developed using galvanic displacement with Co nanoparticles as sacrificial templates, which had a higher surface area and therefore exhibited enhanced electrocatalytic performance.28 Ultrathin PtPdTe and PtTe nanowires were synthesized by using Te nanowires as both sacrificial templates and reducing agents, which induced a higher activity toward the methanol oxidation reaction in comparison with commercial Pt/C.18 For the galvanic displacement of Cu, the replacement of a Cu UPD monolayer by a series of noble metals or their mixtures has been reported for many years, which can only occur on the surface of an electrode. In the bulk preparation, because of the ease for Cu to be oxidized, only a few authors have reported on the galvanic replacement process with Pt. In recent reports, PtCu/C electrocatalysts were prepared by partial galvanic replacement of Cu/C and exhibited high electrocatalytic activity for methanol oxidation29 and oxygen reduction reaction.25 In both reports, PtCu/C was synthesized by a separate two-step procedure. However, the PtCu nanoparticles aggregated severely,25 which would affect the electrochemical surface area and electron transfer.
Herein, PtCu/C catalysts were successfully prepared by galvanic replacement of Cu/C using a successive two-step procedure. This method uses the protection of Ar during the whole reaction process to overcome the issue of Cu oxidation. The dispersion of PtCu nanoparticles was tuned by adjusting the concentration of citric acid with a series of CuCl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :citric acid molar ratios (1:0, 2:1, 1:1, 1:2, 1:5 denoted as PtCu/C-1, PtCu/C-2, PtCu/C-3, PtCu/C-4 and PtCu/C-5, respectively) during the replacement process. All the as-synthesized PtCu/C nanocatalysts exhibited higher activity for methanol oxidation than commercial Pt/C (Pt/C–C) and home-made Pt/C prepared by traditional methods (Pt/C–H), and PtCu/C-3 was evaluated to be the most active catalyst. The X-ray photoelectron spectroscopy (XPS) and electrochemical test detected no unstable Cu atoms on the surface of the metal nanoparticles. This property may provide the as-synthesized PtCu/C catalysts with stability comparable with Pt/C–C and Pt/C–H.
:citric acid molar ratios (1:0, 2:1, 1:1, 1:2, 1:5 denoted as PtCu/C-1, PtCu/C-2, PtCu/C-3, PtCu/C-4 and PtCu/C-5, respectively) during the replacement process. All the as-synthesized PtCu/C nanocatalysts exhibited higher activity for methanol oxidation than commercial Pt/C (Pt/C–C) and home-made Pt/C prepared by traditional methods (Pt/C–H), and PtCu/C-3 was evaluated to be the most active catalyst. The X-ray photoelectron spectroscopy (XPS) and electrochemical test detected no unstable Cu atoms on the surface of the metal nanoparticles. This property may provide the as-synthesized PtCu/C catalysts with stability comparable with Pt/C–C and Pt/C–H.
Fig. 1 shows the TEM images of PtCu/C nanocatalysts, Pt/C–H and Pt/C–C, which exhibit remarkably different morphologies. Due to the low diffraction contrast of Cu, the Cu atoms cannot be seen clearly in the TEM image. However, by careful observation and statistics, it can be seen that Cu nanoparticles with an average size of about 1 nm disperse uniformly on the carbon surface (Fig. 1a). The average size of Pt nanoparticles for Pt/C–C and Pt/C–H are 2.45 nm and 5.0 nm, respectively (Fig. 1b and c). By galvanic displacement, PtCu nanoclusters become ribbon-shaped (Fig. 1d). With the addition of citric acid, the PtCu particles looked like fluffy nanoflowers and the size of the nanoflowers became smaller when the amount of citric acid was increased (Fig. 1e–g). For Fig. 1h, with the molar ratio of CuCl2:citric acid as 1:5, the PtCu nanoclusters became small nanoparticles with uniform size. The shape change of the PtCu nanoclusters can be understood easily. In order to decrease the surface energy, the obtained Pt nanoparticles by galvanic displacement like to attach to each other without the addition of citric acid. However, with the addition of citric acid, the nanoparticles are protected by citric acid and the PtCu nanoclusters become small nanoflowers. The size of the nanoflowers decreased and they finally became nanoparticles when the concentration of citric acid was increased. The metal loading of the catalysts was determined by ICP-MS (Table S1†). From the result and the amount of added precursor, we calculated that almost all the H2PtCl6 was reduced by Cu.
 | ||
| Fig. 1 TEM images of Cu/C (a), Pt/C–C (b), Pt/C–H (c), PtCu/C-1 (d), PtCu/C-2 (e), PtCu/C-3 (f), PtCu/C-4 (g), PtCu/C-5 (h). Scale bars are all 50 nm. | ||
The crystalline structures of the catalysts were characterized by X-ray diffraction (XRD) (Fig. S1†). Each pattern of the catalysts showed a face-centred cubic phase and the unique series of three diffraction peaks for the PtCu/C catalysts had a positive shift compared with Pt/C–C and Pt/C–H, indicating alloy formation in all the PtCu/C catalysts.30,31 XPS was employed to investigate the electronic properties and surface species of the catalyst (Fig. S2†). The Pt 4f peaks for the entire PtCu/C shift negatively compared to that of Pt/C–C and Pt/C–H, which is beneficial to the oxidation of CO. Fig. S2B† shows the Cu 2p region of PtCu/C. There is no characteristic peak of Cu for the catalysts, which indicates that there were no Cu atoms on the surface of the PtCu/C catalysts. The absence of Cu on the surface of PtCu/C can also be demonstrated by the cyclic voltammogram (CV) curves in H2SO4, which have no oxidation peak of Cu in ca. 0.3 V versus SCE (Fig. S3†).
Next, we tested our as-synthesized PtCu/C catalysts in the electrical catalytic reaction and compared the results with those of the as prepared Pt/C–H and Pt/C–C. Fig. 2 details the CV of the catalysts normalized to the mass of Pt. It can be seen that all the PtCu/C catalysts have higher forward oxidation peak current density than Pt/C–H and Pt/C–C, and PtCu/C-3 exhibits the highest peak current density at ca. 3.3 times that of Pt/C–H and Pt/C–C. Chronoamperometry (CA) was performed to investigate the long-term stability of the catalyst for the methanol oxidation reaction (Fig. S4†). Given that the “stability” could be defined as the decay rate of the electrode,32 the current was normalized to the initial current at 100 s to eliminate the difference in the initial current of the catalysts. The residual ratios for Pt/C–C, Pt/C–H, PtCu/C-1, PtCu/C-2, PtCu/C-3, PtCu/C-4 and PtCu/C-5 were 65.6%, 62.2%, 69.6%, 77.8%, 71.0%, 67.8% and 67.9%, respectively, close in value to each other. The lack of a decrease in stability of the PtCu/C catalysts may be attributed to the absence of detectable unstable Cu atoms on the surface of the nanoclusters.
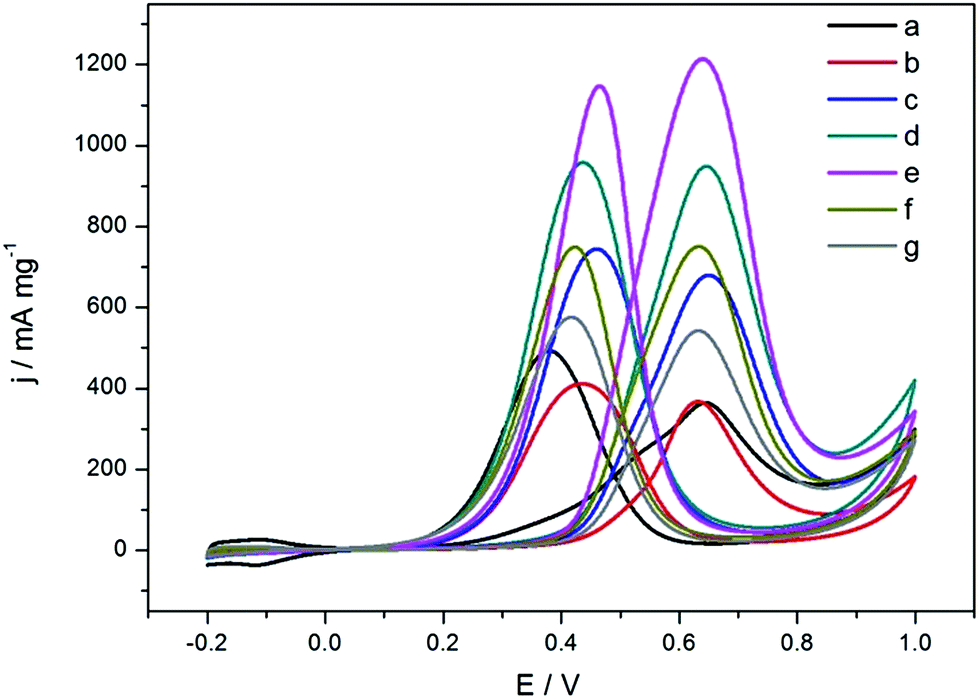 | ||
| Fig. 2 CV of methanol oxidation on Pt/C–C (a), Pt/C–H (b), PtCu/C-1 (c), PtCu/C-2 (d), PtCu/C-3 (e), PtCu/C-4 (f) and PtCu/C-5 (g) in 0.5 M H2SO4 + 1.0 M CH3OH solution at a scan rate of 50 mV s−1. | ||
To explore the reasons for the high catalytic activity of PtCu/C, the CVs for the methanol oxidation with different scan rates of 5, 20, 35, 50, 75, 100, 150 and 200 mV s−1 and a COad stripping experiment of the catalysts were carried out. For comparison, the curves of peak current density against the square root of scan rates of CVs are given in Fig. 3. The obtained linear relationship is attributed to a diffusion-controlled process during the electrooxidation.33 The relationship between the peak current and the square root of the scan rate complies with the following equation in which ip is the peak current density, n is the electron-transfer number for the total reaction, n′, is the electron transfer number in the rate-determining step, α is the electron transfer coefficient of the rate-determining step, A is the electrode surface area, C∝ is the bulk concentration of the reactant, D0 is the diffusion coefficient, ν is the potential scan rate:
| ip = 2.99 × 105 n(αn′)1/2AC∞D1/20ν1/2 |
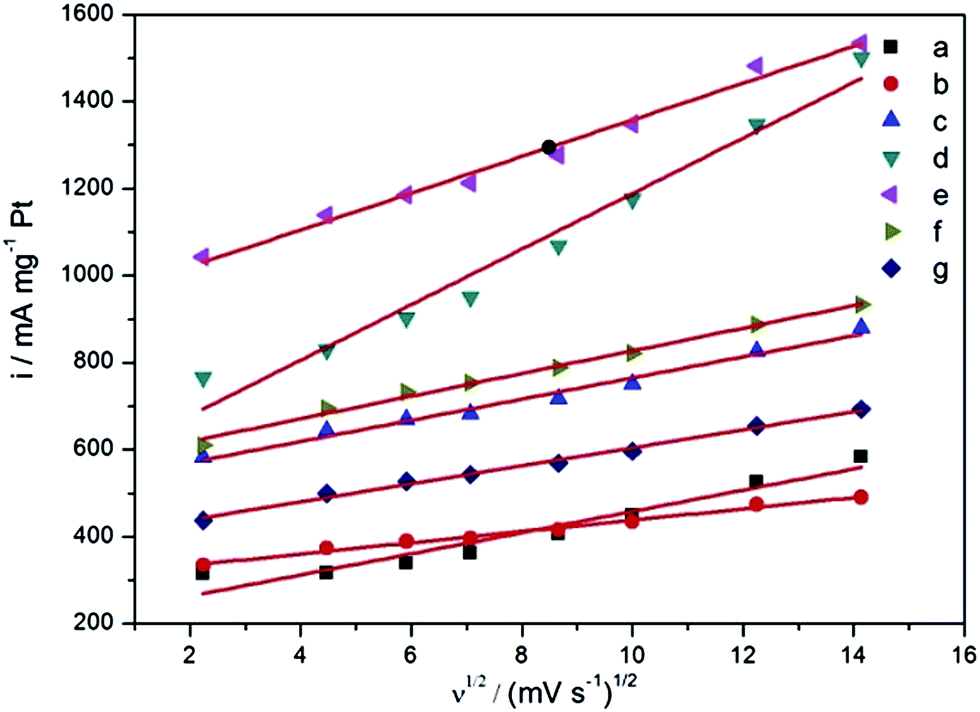 | ||
| Fig. 3 The linear relationship between peak current density and the square root of the scan rates for Pt/C–C (a), Pt/C–H (b), PtCu/C-1 (c), PtCu/C-2 (d), PtCu/C-3 (e), PtCu/C-4 (f) and PtCu/C-5 (g). | ||
In the same electrolyte and the same reaction, the parameters n, C∞ and D0 are the same. Thus, the slop of ip versus the square root of the scan rates is determined by the term of αn′. The corresponding slopes for Pt/C–C, Pt/C–H, PtCu/C-1, PtCu/C-2, PtCu/C-3, PtCu/C-4 and PtCu/C-5 are 24.3, 12.9, 24.3, 63.8, 42.1, 26.0 and 20.7, respectively. This shows that the electron transfer coefficients of these catalysts are in the order of PtCu/C-2 > PtCu/C-3 > PtCu/C-4 > Pt/C–C ≈ PtCu/C-1 > PtCu/C-5 > Pt/C–H. This indicates that the fluffy nanoflower shape for PtCu/C-2, PtCu/C-3 and PtCu/C-4 is greatly advantageous for electron transfer during the methanol oxidation, leading to improved catalytic activity. From the COad stripping curves (Fig. S5†), it can be clearly seen that the peak potentials of COad oxidation on all the PtCu/C catalysts shift to greatly negative compared with that of P/C–C and Pt/C–H. The negative shift indicates the promoted oxidative removal of COad on the PtCu/C catalysts, which is consistent with the inference of XPS. In addition, with the help of citric acid, the electrochemical surface area (ECSA) of PtCu/C-3, PtCu/C-4, and PtCu/C-5 is higher than that of the PtCu/C-1 and PtCu/C-2 catalysts, calculated from the COad stripping voltammetry results (Table S2†), indicating the well-dispersed cluster structure benefits from the exposure of Pt sites. Moreover, the PtCu/C-3 with the appropriate sized fluffy flower structure has a slightly larger ECSA than PtCu/C-1 with a burly nanospheric structure. The large ECSA combined with the electron transfer coefficient gives PtCu/C-3 better catalytic activity for the oxidation of methanol than the other catalysts (Fig. 2).
Conclusions
We have developed a novel synthetic method to obtain dispersion-controlled PtCu/C catalysts by adjusting the concentration of citric acid during the galvanic replacement reaction of Cu to Pt. It was demonstrated that the dispersion has a great effect on the catalytic activity for methanol electrooxidation. Well-dispersed cluster structures benefit from the exposure of Pt sites, electron transfer and COad oxidation. This work can be expected to open the door towards controlling the morphology of metal nanoclusters by the addition of surfactant during the galvanic replacement reaction, allowing for the tuning of their physical-chemical characteristics.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21373199), the National High Technology Research and Development Program of China (863 Program, nos 2012AA053401, 2013AA051002), the National Basic Research Program of China (973 Program, nos 2012CB932800, 2012CB215500 and 2011CB935702), and the Strategic Priority Research Program of the Chinese Academy of Sciences(Grant no. XDA09030104) and the Science & Technology Research Programs of Jilin Province (nos 20102204, 20100420).Notes and references
- Y. Qi, T. Bian, S.-I. Choi, Y. Jiang, C. Jin, M. Fu, H. Zhang and D. Yang, Chem. Commun., 2014, 50, 560–562 RSC.
- D.-M. Gu, Y.-Y. Chu, Z.-B. Wang, Z.-Z. Jiang, G.-P. Yin and Y. Liu, Appl. Catal., B, 2011, 102, 9–18 CrossRef CAS PubMed.
- X. Z. Cui, J. L. Shi, L. X. Zhang, M. L. Ruan and J. H. Gao, Carbon, 2009, 47, 186–194 CrossRef CAS PubMed.
- L. Qian, L. Gu, L. Yang, H. Yuan and D. Xiao, Nanoscale, 2013, 5, 7388–7396 RSC.
- F. Ye, J. Yang, W. Hu, H. Liu, S. Liao, J. Zeng and J. Yang, RSC Adv., 2012, 2, 7479–7486 RSC.
- H.-X. Liu, N. Tian, M. P. Brandon, Z.-Y. Zhou, J.-L. Lin, C. Hardacre, W.-F. Lin and S.-G. Sun, ACS Catal., 2012, 2, 708–715 CrossRef CAS.
- H. Yang, J. Zhang, K. Sun, S. Zou and J. Fang, Angew. Chem., Int. Ed., 2010, 49, 6848–6851 CrossRef CAS PubMed.
- M. Yin, Y. Huang, L. Liang, J. Liao, C. Liu and W. Xing, Chem. Commun., 2011, 47, 8172–8174 RSC.
- Y.-P. Xiao, S. Wan, X. Zhang, J.-S. Hu, Z.-D. Wei and L.-J. Wan, Chem. Commun., 2012, 48, 10331–10333 RSC.
- Y. Tan, J. Fan, G. Chen, N. Zheng and Q. Xie, Chem. Commun., 2011, 47, 11624–11626 RSC.
- J.-H. Jang, J. Kim, Y.-H. Lee, I. Y. Kim, M.-H. Park, C.-W. Yang, S.-J. Hwang and Y.-U. Kwon, Energy Environ. Sci., 2011, 4, 4947–4953 CAS.
- C. Wang, M. F. Chi, D. G. Li, D. Strmcnik, D. van der Vliett, G. F. Wang, V. Komanicky, K. C. Chang, A. P. Paulikas, D. Tripkovic, J. Pearson, K. L. More, N. M. Markovic and V. R. Stamenkovic, J. Am. Chem. Soc., 2011, 133, 14396–14403 CrossRef CAS PubMed.
- Q. Li, P. Xu, B. Zhang, G. Wu, H. Zhao, E. Fu and H.-L. Wang, Nanoscale, 2013, 5, 7397–7402 RSC.
- B. Y. Xia, H. B. Wu, X. Wang and X. W. Lou, J. Am. Chem. Soc., 2012, 134, 13934–13937 CrossRef CAS PubMed.
- R. Loukrakpam, P. Chang, J. Luo, B. Fang, D. Mott, I.-T. Bae, H. R. Naslund, M. H. Engelhard and C.-J. Zhong, Chem. Commun., 2010, 46, 7184–7186 RSC.
- Q. Lv, M. Yin, X. Zhao, C. Li, C. Liu and W. Xing, J. Power Sources, 2012, 218, 93–99 CrossRef CAS PubMed.
- H. J. Qiu and F. X. Zou, ACS Appl. Mater. Interfaces, 2012, 4, 1404–1410 CAS.
- H.-H. Li, S. Zhao, M. Gong, C.-H. Cui, D. He, H.-W. Liang, L. Wu and S.-H. Yu, Angew. Chem., Int. Ed., 2013, 52, 7472–7476 CrossRef CAS PubMed.
- D. Xu, Z. Liu, H. Yang, Q. Liu, J. Zhang, J. Fang, S. Zou and K. Sun, Angew. Chem., Int. Ed., 2009, 48, 4217–4221 CrossRef CAS PubMed.
- J. N. Tiwari, F.-M. Pan and K.-L. Lin, New J. Chem., 2009, 33, 1482–1485 RSC.
- Y. Kang, L. Qi, M. Li, R. E. Diaz, D. Su, R. R. Adzic, E. Stach, J. Li and C. B. Murray, ACS Nano, 2012, 6, 2818–2825 CrossRef CAS PubMed.
- Y. Xu, Y. Yuan, A. Ma, X. Wu, Y. Liu and B. Zhang, ChemPhysChem, 2012, 13, 2601–2609 CrossRef CAS PubMed.
- Y. Xu, S. Hou, Y. Liu, Y. Zhang, H. Wang and B. Zhang, Chem. Commun., 2012, 48, 2665–2667 RSC.
- Y. Xu and B. Zhang, Chem. Soc. Rev., 2014, 43, 2439–2450 RSC.
- B. Geboes, I. Mintsouli, B. Wouters, J. Georgieva, A. Kakaroglou, S. Sotiropoulos, E. Valova, S. Armyanov, A. Hubin and T. Breugelmans, Appl. Catal., B, 2014, 150–151, 249–256 CrossRef CAS PubMed.
- L. Chen, L. Kuai, X. Yu, W. Li and B. Geng, Chem.–Eur. J., 2013, 19, 11753–11758 CrossRef CAS PubMed.
- I. Mintsouli, J. Georgieva, E. Valova, S. Armyanov, A. Kakaroglou, A. Hubin, O. Steenhaut, J. Dille, A. Papaderakis, G. Kokkinidis and S. Sotiropoulos, J. Solid State Electrochem., 2013, 17, 435–443 CrossRef CAS.
- H.-P. Liang, H.-M. Zhang, J.-S. Hu, Y.-G. Guo, L.-J. Wan and C.-L. Bai, Angew. Chem., Int. Ed., 2004, 43, 1540–1543 CrossRef CAS PubMed.
- I. Mintsouli, J. Georgieva, S. Armyanov, E. Valova, G. Avdeev, A. Hubin, O. Steenhaut, J. Dille, D. Tsiplakides, S. Balomenou and S. Sotiropoulos, Appl. Catal., B, 2013, 136, 160–167 CrossRef PubMed.
- X. Z. Cui, J. L. Shi, L. X. Zhang, M. L. Ruan and J. H. Gao, Carbon, 2009, 47, 186–194 CrossRef CAS PubMed.
- A. O. Neto, R. R. Dias, M. M. Tusi, M. Linardi and E. V. Spinace, J. Power Sources, 2007, 166, 87–91 CrossRef PubMed.
- X. Zhao, J. Zhu, L. Liang, J. Liao, C. Liu and W. Xing, J. Mater. Chem., 2012, 22, 19718–19725 RSC.
- M. Yin, Y. Huang, Q. Li, J. O. Jensen, L. N. Cleemann, W. Zhang, N. J. Bjerrum and W. Xing, ChemElectroChem, 2014, 1, 448–454 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: See DOI: 10.1039/c4ra04417g |
| This journal is © The Royal Society of Chemistry 2014 |