
Insights into the catalytic mechanism of dTDP-glucose 4,6-dehydratase from quantum mechanics/molecular mechanics simulations†
Abstract
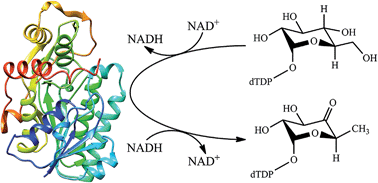
dTDP-glucose 4,6-dehydratase catalyzes the biotransformation of dTDP-glucose into dTDP-4-keto-6-deoxy-glucose. We have utilized the quantum mechanical/molecular mechanical (QM/MM) approach to investigate the detailed mechanism of dTDP-glucose 4,6-dehydratase from Streptomyces venezuelae. On the basis of our calculation results, the previously proposed mechanism has been revised. The overall catalytic cycle can be divided into three sequential chemical steps: oxidation, dehydration and reduction, containing four enzymatic elementary reactions and one non-enzymatic enol–keto tautomerization reaction. The oxidation step proceeds through a concerted asynchronous mechanism with a calculated free energy barrier of 21.1 kcal mol−1, in which the hydride transfer lags behind the proton transfer. The dehydration step prefers a stepwise mechanism rather than a concerted mechanism, and involves an enolate intermediate. Two highly conserved residues Glu129 and Asp128 are involved in this step. In the reduction step, NADH returns the hydride back to glycosyl C6 and the phenolic hydroxyl of Tyr151 spontaneously donates its proton to the C4-keto group, forming an enol sugar as the enzymatic product. After dissociation from the dehydratase active site and diffusion into the solution, this enol sugar will facilely rearrange to give the more favorable dTDP-4-keto-6-dexoyglucose product. Although Thr127 is not directly involved in the whole enzymatic reaction, it is responsible for promoting the catalysis by forming hydrogen-bonding interactions with glycosyl. These calculation results may provide new insight and inspiration for the catalytic mechanism of dTDP-glucose 4,6-dehydratase, even though it is not fully consistent with the previous experimental proposals.
 Please wait while we load your content...
Please wait while we load your content...

