
Highly selective catalytic hydrodeoxygenation of Caromatic–OH in bio-oil to cycloalkanes on a Ce–Ni–W–B amorphous catalyst
Abstract
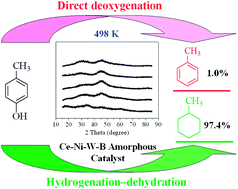
This study focused on the preparation of Ce–Ni–W–B amorphous catalysts and the effect of Ce content on their catalytic activities in the hydrodeoxygenation (HDO) of phenols in bio-oil. Adding the promoter Ce could increase the content of Ni0 and WO3 on the Ce–Ni–W–B catalyst surface, leading to the improvement of the deoxygenation activity, but excess Ce would cover some active sites, resulting in a reduction of the catalytic activity. Because of the amorphous structure and the electron transfer between Ni0 and B0, these catalysts possess very high hydrogenation activity, making the HDO of phenols on these amorphous catalysts proceed with a hydrogenation-dehydration route, which not only decreases the aromatic content in the product but also the reaction temperature. With an optimal Ce content (2.5 mol%), the total aromatic selectivity reduced to 1.0% and the HDO reaction temperature decreased to 498 K. This research provides a high activity catalyst for transforming phenols into cycloalkanes.
 Please wait while we load your content...
Please wait while we load your content...

