
Impact of serum proteins on MRI contrast agents: cellular binding and T2 relaxation†
Abstract
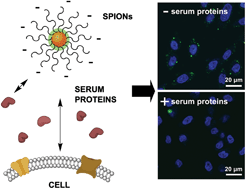
Superparamagnetic iron oxide nanoparticles (SPIONs) used as MRI contrast agents or for theranostic applications encounter a complex mixture of extracellular proteins that adsorb on the SPION surface forming a protein corona. Our goal was to understand how cellular binding and T2 relaxation times are affected by this protein corona. Our studies focused on carboxymethyl dextran-modified SPIONs, chosen for their similarity to Resovist SPIONs used to detect liver lesions. Using a combination of fluorescence microscopy and flow cytometry, we find that the cellular binding of SPIONs to both macrophages and epithelial cells is significantly inhibited by serum proteins. To determine if this decreased binding is due to the iron oxide core or the carboxymethyl dextran surface coating, we functionalized polystyrene nanoparticles with a similar carboxymethyl dextran coating. We find a comparable decrease in cellular binding for the carboxymethyl dextran–polystyrene nanoparticles indicating that the carbohydrate surface modification is the key factor in SPION–cell interactions. NMR measurements showed that T2 relaxation times are not affected by corona formation. These results indicate that SPIONs have a decreased binding to cells under physiological conditions, possibly limiting their use in theranostic applications. We expect these results will be useful in the design of SPIONs for future diagnostic and therapeutic applications.
 Please wait while we load your content...
Please wait while we load your content...

