
Combinatorial bio-conjugation of gemcitabine and curcumin enables dual drug delivery with synergistic anticancer efficacy and reduced toxicity†
Abstract
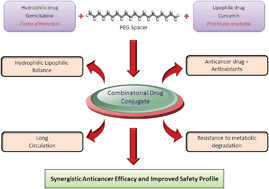
The present study seeks to exploit a novel bio-conjugation strategy for improving the biopharmaceutical properties of gemcitabine (GEM) while enhancing its anticancer efficacy. A macromolecular bio-conjugate comprised of GEM and curcumin (CUR), was synthesized and authenticated. The effect of bio-conjugation was estimated on the physicochemical properties and stability in different buffers and plasma. MTT assay on human breast adenocarcinoma MCF-7 cell lines, apoptosis assay and DNA damage assay were performed to access the in vitro cell cytotoxicity. The conjugate was further tested in vivo for tumor growth inhibition studies in DMBA breast cancer induced SD rats and toxicity in Swiss mice. Covalent conjugation of GEM with CUR via a PEG spacer transformed the solubility profile of GEM–PEG–CUR and significantly improved the stability in aqueous buffers and plasma. GEM–PEG–CUR exhibited significantly higher cell cytotoxicity in comparison with free drug congeners (viz. free CUR/GEM), their combination and PEGylated congeners. Significantly higher tumor growth inhibition and lower toxicity further established the superiority of GEM–PEG–CUR conjugate over other pharmaceutical preparations in terms of both efficacy and safety. The results clearly indicate that the dual drug conjugation is an effective means to synergize the therapeutic potential of drug candidates while alleviating drug-associated toxicity.
 Please wait while we load your content...
Please wait while we load your content...

