
Is metal metathesis a framework-templating strategy to synthesize coordination polymers (CPs)? Transmetallation studies involving flexible ligands†
Abstract
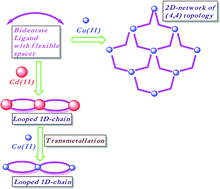
The coordination polymer (CP) of bis(3-pyridyl)butanediamide with Cu(ClO4)2 is a 2D network (CP-3-Cu), which showed two-fold parallel interpenetration, whereas with Cd(ClO4)2 it is a 1D network containing rectangular loops (CP-4-Cd). The metal metathesis of CP-4-Cd with Cu(II) resulted in the isomorphous replacement of the Cd(II) centre with Cu(II). Transmetallation reaction resulted in retaining the structural features of CP-4-Cd even in case of a flexible ligand. The CP formed via transmetallation couldn't be synthesized from a direct reaction of bis(3-pyridyl)butanediamide and Cu(ClO4)2. The transmetallation kinetic studies were performed with an atomic absorption spectrophotometer (AAS) and wavelength dispersive X-ray fluorescence (WDXRF).
 Please wait while we load your content...
Please wait while we load your content...

