
Highly efficient homogeneous and heterogenized ruthenium catalysts for transfer hydrogenation of carbonyl compounds†
Abstract
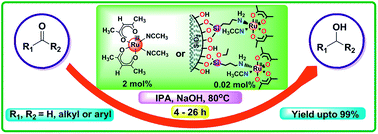
[Ru(acac)2(CH3CN)2] was synthesized from [Ru(acac)3] by refluxing it with zinc powder in ethanol–acetonitrile mixture. The prepared catalyst was characterized by FT-IR, 1H and 13C NMR techniques. The silica supported catalyst (‘SiO2’–NH2–RuII) was prepared by stirring [Ru(acac)2(CH3CN)2] with ‘SiO2’–NH2 in a 1 : 1 mixture of CH2Cl2–toluene for 3 days at room temperature. It was characterized by FT-IR, SEM, solid state NMR, ICP and BET surface area methods. The transfer hydrogenation reaction of carbonyl compounds was carried out separately using [Ru(acac)2(CH3CN)2] and ‘SiO2’–NH2–RuII as catalysts. The reaction conditions were optimized with different solvents, bases, catalyst amounts and temperatures using acetophenone as a model system. The scope of the reaction was extended to various substituted aryl ketones including heteroaryl ketones.
 Please wait while we load your content...
Please wait while we load your content...

