
Mesoporous carbon coated molybdenum oxide nanobelts for improved lithium ion storage†
Abstract
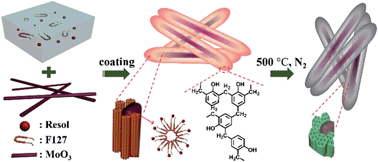
Nanocomposites composed of mesoporous carbon coated molybdenum oxide nanobelts are prepared and used as anode materials for Li-ion batteries. The obtained MoOx/meso-C nanocomposites provide a high surface area for electrochemical reaction, large mesopore channels for lithium ion transport, improved electrical conductivity, and structural flexibility for electrode volume change.
 Please wait while we load your content...
Please wait while we load your content...

