
Nitroalkenes in the synthesis of carbocyclic compounds
Abstract
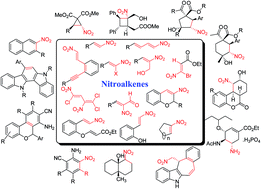
The applications of nitroalkenes in the synthesis of small, common and medium ring carbocycles, including natural products are investigated in this review. These carbocyclic compounds were synthesized from cyclic or acyclic nitroalkenes via a wide variety of reactions such as Michael addition, Diels–Alder reaction, 1,3-dipolar and cycloaddition, Morita–Baylis–Hillman reaction and many cascade reactions often with high regio- and stereoselectivities. Nitroalkenes with a variety of substitution patterns including electroneutral, electron donating and electron withdrawing groups at α- and/or β-positions are suitable substrates for the synthesis of the carbocyclic compounds. The high reactivity of nitroalkenes and their ability to coordinate the metal catalysts as well as organocatalysts signify them as efficient substrates in synthetic organic chemistry.
 Please wait while we load your content...
Please wait while we load your content...

