
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
One-pot route to β-adrenergic blockers via enantioselective organocatalysed epoxidation of terminal alkenes as a key step†
Felix E.
Held
,
Shengwei
Wei
,
Kathrin
Eder
and
Svetlana B.
Tsogoeva
*
Department of Chemistry and Pharmacy, Chair of Organic Chemistry I and Interdisciplinary Center for Molecular Materials (ICMM), University of Erlangen-Nürnberg, Henkestrasse 42, 91054 Erlangen, Germany. E-mail: svetlana.tsogoeva@fau.fe; Fax: +49-9131-85-26865
First published on 16th July 2014
Abstract
A convenient and environmentally attractive one-pot two-step process for the synthesis of β-adrenergic blockers via Shi's organocatalytic epoxidation of terminal alkenes and subsequent aminolysis reaction of epoxides with isopropylamine under mild reaction conditions has been developed.
Introduction
The synthesis of enantiomerically pure chiral drugs and bioactive compounds still remains a challenging task for organic chemists.Asymmetric syntheses employing chiral catalysts is among the most common routes to optically pure compounds. In particular, organocatalysis, which constitutes a complimentary extension to transition metal or enzymatic catalysis, has attracted much attention in recent years as a powerful, and environmentally friendly methodology.1
Most of the known organocatalytic synthesis are carried out through the accustomed classical stop-and-go approach, where intermediate isolation and purification steps are involved. In general, isolation and purification processes of intermediates are the most time- and cost-demanding and waste-producing manual operations in the traditional stop-and-go synthetic approach.2
In contrast to this approach stands the one-pot strategy, in which chemical transformations are performed without intermediary purification steps. One-pot reactions are therefore among the economically most attractive and sustainable methods in modern synthetic chemistry.2–5
Chiral 1,2-amino alcohols are versatile building blocks in the synthesis of biologically active target compounds and are subunits in many natural products and therapeutic agents, such as bronchodilators, neurotransmitters, sympathomimetic agents, and many others.6 1,2-Amino alcohols represent, therefore, attractive synthetic targets.
An example of two-step epoxide synthesis employing oxathiane as a chiral reagent and application of the isolated epoxides to preparation of bioactive 1,2-amino alcohols via aminolysis reaction with LiAl(NHR)4/NH2R was reported in 1995.7
In 2011, we introduced a one-pot multi-step synthetic approach towards 1,2-amino alcohols employing oxazaborolidine organocatalysts, prepared in situ from α-amino acids.8 This new strategy we applied for the highly enantioselective synthesis of selected β-adrenergic blockers 1–3 (Fig. 1) via one-pot sequential transformations: reduction of α-halo ketones/epoxide formation/aminolysis reaction.8 Pharmaceuticals pronethalol (1),9 nifenalol (2)10 and dichloroisoproterenol (3)11 are of great importance in the therapy of asthma, bronchitis and congestive heart failure.12
 | ||
| Fig. 1 Selected β-adrenergic blockers. | ||
Very recently, a new method for the preparation of (R)-pronethalol (1) and (R)-dichloroisoproterenol (3) in moderate yields and high enantioselectivities (41%, 96% ee and 60%, 98.6% ee, respectively) was developed, which involves acetylcyanation of prochiral aldehydes catalysed by a combination Ti-based Lewis acid catalyst and a biocatalyst employing a minor enantiomer recycling procedure, followed by a subsequent LAH or BH3 reduction, respectively, and reductive amination using NaBH4/acetone.13
Some practical shortcomings of the previously reported synthetic methodologies towards selected pharmaceuticals includes the involvement of multiple reaction steps,7,8,13 the requirement of isolation and purification of product intermediates7,13 and the use of a relatively complex catalytic system (a combination of metal catalyst and biocatalyst)13 and Li-based reagents (LiAl(NHR)4, LAH).7,13 To match the basic needs of the pharmaceutical chemistry, the synthetic methods towards chiral drugs must be simple, straightforward and environmentally friendly. Thus, finding a shorter, facile and metal-free strategy providing a more convenient direct access to these chiral β-adrenergic antagonists under mild reaction conditions and without intermediary workup and purification steps, further reducing costs and waste, is very welcome.
Here we report a significant progress towards this goal with the development of a simple and practical organocatalytic epoxidation/aminolysis sequential one-pot route to the synthesis of β-adrenergic blockers (Fig. 2).
 | ||
| Fig. 2 Depiction of the designed short enantioselective synthesis of chiral 1,2-amino alcohols. | ||
Results and discussion
From a synthetic perspective, epoxides are particularly attractive and versatile starting compounds in organic synthesis and in the pharmaceutical industry.14,15For realisation of the proposed synthetic route (Fig. 2), asymmetric epoxidation utilising organocatalysts has attracted our attention. Epoxidation of styrenes (selected here as a key reaction step) is still a challenging subject in the field of oxidation chemistry. An attractive metal-free method described for the epoxidation of terminal alkenes is the Shi epoxidation.16,17 However, as far as we are aware, this method has never been applied to the synthesis of β-adrenergic blockers. We envisaged that employing versatile Shi epoxidation reaction as a key step to build up selected β-adrenergic blockers 1–3 in a one-pot process would be of synthetic value.
The Shi's N-substituted oxazolidinone ketone 4 (Fig. 3) has proven to be one of the most active types of fructose-derived asymmetric organocatalysts for epoxidation of unfunctionalised terminal alkenes.
 | ||
| Fig. 3 Shi catalyst applied for epoxidation step. | ||
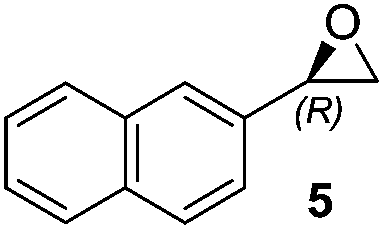
Initially, we examined the epoxidation of 2-vinylnaphthalene in the presence commercially available N-substituted oxazolidinone ketone 4 using three conventional solvents (n-hexane, CH3CN and THF) different from DME (dimethoxyethane) originally reported for Shi epoxidation reaction.17
The laboratory work with toxic DME is known to imply several risks, for instance it may impair fertility and furthermore cause fetal harm.18 Since the β-blockers 1–3 are administered as drugs it would be very desirable to find a comparable solvent without any loss of conversion and enantiomeric excess but at the same time with a less harmful effect.
Hence, it was our motivation first to carry out preliminary solvent screening in the epoxidation reaction step (Table 1). The epoxidation of 2-vinylnaphthalene to epoxide 5 was performed at 0 °C, beginning with 4.0 equiv. of Oxone® as an oxidizing agent. Both, the catalytic efficiency and asymmetric induction are strongly dependent on the solvents used. In n-hexane and acetonitrile, for instance, the reaction didn't work at all or only with a low conversion, respectively (entries 1 and 2). To our delight, changing the reaction medium to THF, we observed a high conversion of 94% with an enantiomeric excess of 82% after only 6 hours of reaction time (entry 3). Furthermore, THF offers another big advantage for the whole reaction system, since previously, our group identified this solvent as the most suitable for the following aminolysis reaction step.8
|
|
||||||
|---|---|---|---|---|---|---|
| Entrya | Product | Oxone®f (equiv.) | Solvent | t (h) | conv.b (%) | eec (%) |
| a All reactions were carried out with Shi cat. 4 (0.15 equiv.), Oxone® (2.6–4.0 equiv.), and K2CO3 (4 equiv., 0.84 M in 4 × 10−4 M aq. EDTA) and Bu4NHSO4 (0.09 equiv.) in different solvents and buffer (0.1 M K2CO3–AcOH in 4 × 10−4 M aq. EDTA, pH = 9.3) unless otherwise stated. b Determined by 1H-NMR. c Determined by chiral phase HPLC analysis and compared with authentic racemic material. The absolute configurations were assigned by comparing HPLC elution order with known literature data. d Not determined. e The reaction was carried out at r.t. f Oxone®: (2KHSO5·KHSO4·K2SO4). | ||||||
| 1 |
|
4.0 | n-hexane | 8 | Traces | n.d.d |
| 2 | 4.0 | CH3CN | 8 | 25 | 79 | |
| 3 | 4.0 | THF | 6 | 94 | 82 | |
| 4 | 2.6 | THF | 6 | >99 | 83 | |
| 5e | 2.6 | THF | 6 | 82 | 65 | |
| 6 | 2.6 | THF | 8 | >99 | 82 | |
| 7 | 2.6 | THF | 4 | 90 | 87 | |
| 8 |
|
2.6 | THF | 6 | >99 | 81 |
| 9 |

|
2.6 | THF | 6 | >95 | 84 |

For further reaction optimization, the amounts of the oxidizing agent was reduced to 2.6 equiv. Carrying out the reaction for 6 h in THF at 0 °C the product was generated with >99% conversion and 83% ee (entry 4, Table 1). To investigate the influence of the temperature on the reaction outcome, we carried out the epoxidation also at room temperature. This experiment gave evidence that an elevated temperature affected the conversion and enantioselectivity in a negative way (entry 5), possibly caused by a decomposition of the used catalyst during the reaction course.19 Among the different reaction times that were applied for the epoxidation, an extended reaction period (8 h, entry 6) didn't have any significant effect on the reaction outcome, while a shortened period (4 h, entry 7) reduced the conversion to 90%. Thus, 6 h reaction time proved to be the most optimal for the epoxidation step.
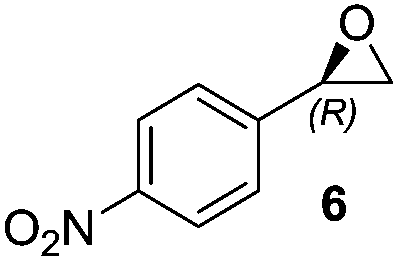
Thus, improved conditions to those previously reported17 for the Shi epoxidation of terminal alkenes, suitable for designed one-pot transformation (Fig. 1), were found. We have next applied this optimized method for the synthesis of epoxides 6 and 7, which are ideal precursors for (R)-nifenalol (2) and (R)-dichloroisoproterenol (3) synthesis, respectively. After 6 h reaction time both epoxides were obtained with excellent to full conversion and enantiomeric ratios of 81 and 84%, respectively (entries 8 and 9).
The next aim was to combine this epoxidation reaction (without work up of the terminal epoxide) with the second aminolysis reaction step in a one-pot process (Table 2).
|
|
|||
|---|---|---|---|
| Entrya | Products | Yieldb (%) over two steps | eec (%) |
| a All reactions were carried out with Shi cat. 4 (0.15 equiv.), Oxone® (4.0 equiv.), and K2CO3 (4 equiv., 0.84 M in 4 × 10−4 M aq. EDTA) and Bu4NHSO4 (0.09 equiv.) in THF and buffer (0.1 M K2CO3–AcOH in 4 × 10−4 M aq. EDTA, pH = 9.3) unless otherwise stated. b Obtained via recrystallization. c Determined by chiral phase HPLC analysis and compared with authentic racemic material after isolation via preparative TLC. The absolute configuration was established by comparing HPLC elution order with known literature data.8 d ee-value after a single recrystallization. e Single crystal for the X-ray diffraction analysis of the (R)-pronethalol (1) was obtained and the structure was unambiguously determined by X-ray crystallography (see the ESI†).20 f Not determined. g DME was used as solvent. h Carried out at −10 °C. i Oxone®: (2KHSO5·KHSO4·K2SO4). | |||
| 1 |

|
n.d | 89 (99)d |
| 2 | 55 | 81 | |
| 3 | n.d.f | 90 (99)d,e | |
| 4 | n.d.f | 84 | |
| 5 | n.d.f | 87 | |
| 6g | 44 | 84 (99)d | |
| 7g,h | 59 | 87 | |
| 8 |

|
n.d.f | 84 |
| 9 | n.d.f | 82 | |
| 10 | 60 | 85 (93)d | |
| 11 |

|
n.d.f | >99d |
| 12 | 78 | 83 (99)d | |
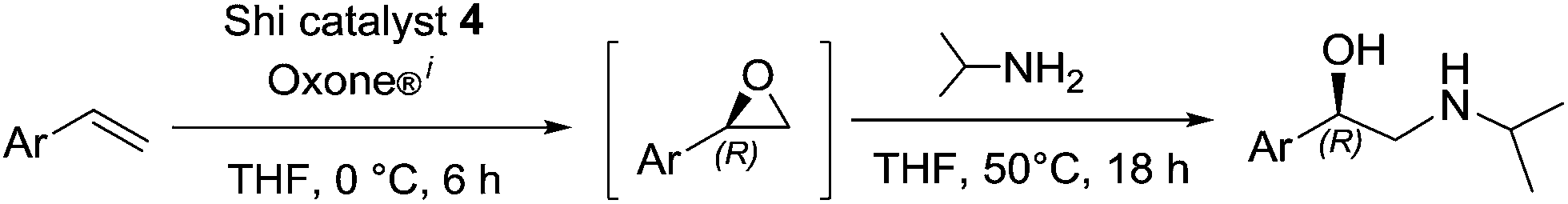
We started with the development of the one-pot process for (R)-pronethalol (1) synthesis. For the epoxidation reaction, Oxone® and K2CO3 were added in a buffer solution dropwise simultaneously via two syringe pumps in order to ensure an ideal performance of the Shi catalyst 4 with a constant pH value during the epoxidation. After 6 h reaction time in THF we added 18 equiv. of isopropylamine to the reaction mixture and stirred further at 50 °C for 18 h. We used 18 equiv. of isopropylamine, since our previous studies8 demonstrated that an excess of isopropylamine is crucial for the aminolysis step for a complete consumption of epoxide.
(R)-pronethalol (1) was isolated in 55% yield after two sequential steps (entry 2). Enantiomeric ratios were reproducibly high (up to 90%, entries 1–5). By recrystallization, we further improved the ee-value of the product with respect to the value determined directly from the crude reaction mixture. Crystals were obtained in an almost enantiopure quality (up to 99% ee, entries 1 and 3) after single recrystallization.
In addition, we carried out the first step of the one-pot reaction under original conditions, applied by Shi et al. for the epoxidation reaction in DME at −10 °C,17 and were delighted to find that the catalyst 4 performed even better in THF at 0 °C than in DME at 0 °C or −10 °C (entries 1–5 vs. entries 6 and 7). Thus, within our current research, we found that THF was a more suitable solvent than DME for the one-pot reaction system with respect to both the yield and the enantiomeric ratio.
Subsequently, we also applied the developed one-pot reaction route to the synthesis of two additional target compounds (R)-nifenalol (2) and (R)-dichloroisoproterenol (3). The corresponding products 2 and 3 were obtained with 60% and 78% yields after two sequential steps, respectively (entries 10 and 12). Regarding the enantiocontrol of these reactions, we gained similar good results as compared to the (R)-pronethalol synthesis (entries 8–12 versus entries 1–5).
It is indicated, that the prepared β-blockers crystallize as conglomerates, since on recrystallization of 2 and 3, we again observed an enantioenrichment in the products as the obtained crystals had a significantly higher ee-value (93% and 99%, respectively) as compared to the dissolved products in the crude reaction mixture (85% and 83%, entries 10 and 12, respectively).
Conclusion
In summary, a new one-pot process providing a convenient two-step route for the formation of β-blockers: (R)-pronethalol (1), (R)-nifenalol (2) and (R)-dichloroisoproterenol (3), from corresponding terminal alkenes was developed.While the individual reactions selected for realisation of the one-pot synthesis (Shi-epoxidation and aminolysis reaction) are already known, the combination of these transformations in a one-pot two-step process leading to chiral drugs is unprecedented.
The use of commercially available Shi's N-substituted oxazolidinone ketone 4 as an organocatalyst for the epoxidation of terminal alkenes, followed by the aminolysis reaction with isopropylamine, and the mild reaction conditions used for both transformations, make this one-pot two-step process – up to the present – the most simple, shortest and environmentally friendly approach to obtain β-blockers.
Experimental
General information
All reagents purchased from Sigma-Aldrich Co. or Acros Organics were used without further purification. n-Hexane, CH3CN, THF (HPLC grade) and DME was obtained from Fisher Scientific and used as received. Column chromatography was performed on Acros silica gel 60 Å and TLC on Macherey-Nagel ALUGram® SIL G/UV254 plates. 1H NMR and 13C NMR spectra were recorded on Bruker Avance 400 or Bruker Avance 300 NMR spectrometers using the solvent as internal standard. The enantiomeric excess of products was determined by chiral HPLC analysis in comparison with authentic racemic material. HPLC measurements were performed using Agilent Technologies 1200 Series equipment.General procedure for the asymmetric epoxidation
To a solution of the corresponding alkene (0.20 mmol, 1 equiv.) and Shi catalyst 4 (0.010 g, 0.030 mmol, 0.15 equiv.) in THF (3.0 mL) were added buffer (0.1 M K2CO3–AcOH in 4 × 10−4 M aq. EDTA, pH = 9.3, 2.0 mL) and Bu4NHSO4 (0.0060 g, 0.018 mmol, 0.09 equiv.) with stirring. After the mixture was cooled to 0 °C (bath temperature) via an ice bath, a solution of Oxone® (0.33 g, 0.53 mmol, 2.6 equiv.) in 4 × 10−4 M aq. EDTA (2.5 mL) and a solution of K2CO3 (0.84 M in 4 × 10−4 M aq. EDTA, 2.5 mL) were added dropwise simultaneously within 6 h via syringe pumps. The reaction was quenched by dilution with DCM and extracted with DCM. The combined organic layers were dried over Na2SO4, concentrated, and purified by flash chromatography (SiO2, petroleum ether–ethyl acetate) to give the corresponding epoxides as a colorless solid.2-(Naphthalen-2-yl)oxirane (5)21
1H NMR (300 MHz, CDCl3): δ = 7.82–7.89 (m, 4H), 7.49–7.45 (m, 2H), 7.31 (dd, 1H, J = 1.6 Hz, and 8.4 Hz), 4.02 (dd, 1H, J = 2.8 Hz, and 4.0 Hz), 3.21 (dd, 1H, J = 4.0 Hz, and 5.2 Hz), 2.90 (dd, 1H, J = 2.4 Hz, and 5.6 Hz). Chiral HPLC (Chiralpak IC, n-hexane–2-PrOH 99![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, flow rate 0.5 mL min−1, λ = 254 nm): tR 18.2 min (R), 21.5 min (S).
:1, flow rate 0.5 mL min−1, λ = 254 nm): tR 18.2 min (R), 21.5 min (S).
2-(4-Nitrophenyl)oxirane (6)8,22
1H NMR (300 MHz, CDCl3): δ = 8.19 (d, 2H, J = 8.8 Hz), 7.43 (d, 2H, J = 8.7 Hz), 3.94 (dd, 1H, J = 2.4 Hz, and 4.0 Hz), 3.21 (dd, 1H, J = 4.2 Hz, and 5.5 Hz), 2.76 (dd, 1H, J = 2.4 Hz, and 5.5 Hz). Chiral HPLC (Chiralpak IC, n-hexane–2-PrOH 95:5, flow rate 1 mL min−1, λ = 254 nm): tR 13.9 min (R), 14.8 min (S).
(3,4-Dichlorophenyl)oxirane (7)23
1H NMR (300 MHz, CDCl3): δ = 7.39 (d, 2H, J = 8.2 Hz), 7.33 (d, 1H, J = 2.0 Hz), 7.09 (dd, 1H, J = 2.0 Hz, and 8.3 Hz), 3.79 (dd, 1H, J = 2.6 Hz, and 3.9 Hz), 3.12 (dd, 1H, J = 4.1 Hz, and 5.4 Hz), 2.71 (dd, 1H, J = 2.5 Hz, and 5.6 Hz). Chiral HPLC (Chiralpak IC, n-hexane–2-PrOH 98:2, flow rate 1 mL min−1, λ = 280 nm): tR 6.1 min (R), 7.2 min (S).
General procedure for the one-pot two-step synthesis of β-blockers
To a solution of the corresponding alkene (0.20 mmol, 1 equiv.) and Shi catalyst 4 (0.010 g, 0.030 mmol, 0.15 equiv.) in THF (3.0 mL) were added buffer (0.1 M K2CO3–AcOH in 4 × 10−4 M aq. EDTA, pH = 9.3, 2.0 mL) and Bu4NHSO4 (0.0060 g, 0.018 mmol, 0.09 equiv.) with stirring. After the mixture was cooled to 0 °C (bath temperature) via an ice bath, a solution of Oxone® (0.33 g, 0.53 mmol, 2.6 equiv.) in 4 × 10−4 M aq. EDTA (2.5 mL) and a solution of K2CO3 (0.84 M in 4 × 10−4 M aq. EDTA, 2.5 mL) were added dropwise simultaneously within 6 h via syringe pumps. To this mixture isopropylamine (0.30 mL, 3.5 mmol, 18 equiv.) was added and the stirring was continued at 50 °C for 18 hours. The organic solvent was evaporated and the remaining aqueous layer was extracted with ethyl acetate (2 × 15 mL). The combined organic layers were dried over Na2SO4. The solvent was removed under reduced pressure and the residue was recrystallized from ethyl acetate–n-hexane to give the corresponding β-blockers as colorless crystals.(R)-Pronethalol (1)8,24
White solid. 1H NMR (300 MHz, CDCl3): δ = 7.80–7.83 (m, 4H), 7.43–7.47 (m, 3H), 4.81 (dd, 1H, J = 3.68, and 8.60 Hz), 3.01 (dd, 1H, J = 3.75 Hz, and 12.07 Hz), 2.83 (sept, 1H, J = 6.26 Hz), 2.72 (dd, 1H, J = 8.68 Hz, and 12.07 Hz), 1.07 (dd, 6H, J = 1.85 Hz, and 6.24 Hz). 13C-NMR (300 MHz, CDCl3): δ = 139.9, 132.8, 132.5, 127.7, 127.5, 127.2, 126.6, 125.3, 124.1, 123.6, 71.6, 54.1, 48.3, 22.7, 22.5. Chiral HPLC (Chiralpak IC, n-hexane–2-PrOH–ethanolamine 94.9:5:0.1, flow rate 1 mL min−1, λ = 280 nm): tR 8.3 min (R), 10.4 min (S).
(R)-Nifenalol (2)8,24
White solid. 1H NMR (300 MHz, CDCl3): δ = 8.16 (d, 2H, J = 8.4 Hz), 7.51 (d, 2H, J = 8.5 Hz), 4.82 (dd, 1H, J = 3.2, and 9.2 Hz), 2.96 (dd, 1H, J = 3.4, and 12.2 Hz), 2.40–2.64 (m, 1H), 2.60 (dd, 1H, J = 9.3, and 12.2 Hz), 1.11 (d, 3H, J = 6.7 Hz), 1.10 (d, 3H, J = 6.7 Hz). 13C-NMR (300 MHz, CDCl3): δ = 149.9, 146.8, 126.0, 123.2, 70.4, 53.7, 48.4, 22.7, 22.4. MS (EI) m/z (%): 151 (5), 72 (100). Chiral HPLC (Chiralpak IC, n-hexane–2-PrOH–triethylamine 94.9:5:0.1, flow rate 0.5 mL min−1, λ = 280 nm): tR 25.1 min (R), 26.5 min (S).
(R)-Dichloroisoproterenol (3)8,24
White solid. 1H NMR (300 MHz, CDCl3): δ = 7.44 (d, 1H, J = 1.88 Hz), 7.37 (d, 1H, J = 8.26 Hz), 7.15 (dd, 1H, J = 1.62 Hz, and 8.28 Hz), 4.57 (dd, 1H, J = 3.61 Hz, and 9.01 Hz), 2.86 (dd, 1H, J = 3.64 Hz, and 12.12 Hz), 2.77 (sept, 1H, J = 6.29 Hz), 2.54 (dd, 1H, J = 9.03, and 12.13 Hz), 1.04 (d, 6H, J = 6.28 Hz). 13C-NMR (300 MHz, CDCl3): δ = 143.2, 132.4, 131.1, 130.2, 127.7, 125.1, 70.7, 54.3, 48.7, 23.1, 22.9. MS (EI) m/z: 230 (M–OH, 13%), 214 (M–Cl, 100). Chiral HPLC (Chiralpak IC, n-hexane–2-PrOH–ethanolamine 94.9:5:0.1, flow rate 0.5 mL min−1, λ = 280 nm): tR 10.3 min (R), 11.1 min (S).
Acknowledgements
We thank Dr Frank Hampel, Institute of Organic Chemistry I, for the X-Ray structure determination. S. B. T. is grateful to the Interdisciplinary Center for Molecular Materials (ICMM) and Erlangen Catalysis Resource Center (ECRC) for research support.Notes and references
- For general reviews on asymmetric organocatalysis, see: (a) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2001, 40, 3726 CrossRef CAS; (b) P. I. Dalko and L. Moisan, Angew. Chem., Int. Ed., 2004, 43, 5138 CrossRef CAS PubMed; (c) K. N. Houk and B. List, Acc. Chem. Res., 2004, 37, 487 CrossRef CAS; (d) A. Berkessel and H. Gröger, Asymmetric Organocatalysis, Wiley-VCH, Weinheim, 2005 Search PubMed; (e) B. List and J. W. Yang, Science, 2006, 313, 1584 CrossRef CAS PubMed; (f) Enantioselective Organocatalysis, ed. P. I. Dalko, Wiley-VCH, Weinheim, 2007 Search PubMed; (g) H. Pellissier, Tetrahedron, 2007, 63, 9267 CrossRef CAS PubMed; (h) A. Dondoni and A. Massi, Angew. Chem., Int. Ed., 2008, 47, 4638 CrossRef CAS PubMed; (i) P. Melchiorre, M. Marigo, A. Carlone and G. Bartoli, Angew. Chem., Int. Ed., 2008, 47, 6138 CrossRef CAS PubMed.
- Ł. Albrecht, H. Jiang and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 8492 CrossRef PubMed and references therein.
- (a) K. C. Nicolaou, T. Montagnon and S. A. Snyder, Chem. Commun., 2003, 551 RSC; (b) L. F. Tietze, G. Brasche and K. Gericke, Domino Reactions in Organic Synthesis, Wiley-VCH, Weinheim, 2006 Search PubMed; (c) K. C. Nicolaou, D. J. Edmonds and P. G. Bulger, Angew. Chem., 2006, 118, 7292 ( Angew. Chem., Int. Ed. , 2006 , 45 , 7134 ) CrossRef; (d) D. Enders, C. Grondal and M. R. M. Hüttl, Angew. Chem., 2007, 119, 1590 ( Angew. Chem., Int. Ed. , 2007 , 46 , 1570 ) CrossRef; (e) C. Vaxelaire, P. Winter and M. Christmann, Angew. Chem., Int. Ed., 2011, 50, 3605 CrossRef CAS PubMed.
- For selected examples, see: (a) Y. Huang, A. M. Walji, C. H. Larsen and D. W. C. MacMillan, J. Am. Chem. Soc., 2005, 127, 15051 CrossRef CAS PubMed; (b) D. Enders, M. R. M. Hüttl, J. Runsink, G. Raabe and B. Wendt, Angew. Chem., 2007, 119, 471 ( Angew. Chem., Int. Ed. , 2007 , 46 , 467 ) CrossRef; (c) G.-L. Zhao, R. Rios, J. Vesely, L. Eriksson and A. Córdova, Angew. Chem., 2008, 120, 8596 ( Angew. Chem., Int. Ed. , 2008 , 47 , 8468 ) CrossRef; (d) S. Belot, K. A. Vogt, C. Besnard, N. Krause and A. Alexakis, Angew. Chem., 2009, 121, 9085 ( Angew. Chem., Int. Ed. , 2009 , 48 , 8923 ) CrossRef; (e) H. Jiang, N. Holub, M. W. Paixão, C. Tiberi, A. Falcicchio and K. A. Jørgensen, Chem.–Eur. J., 2009, 15, 9638 CrossRef CAS PubMed; (f) C. Chandler, P. Galzerano, A. Michrowska and B. List, Angew. Chem., 2009, 121, 2012 ( Angew. Chem., Int. Ed. , 2009 , 48 , 1978 ) CrossRef; (g) C. Grondal, M. Jeanty and D. Enders, Nat. Chem., 2010, 2, 167 CrossRef CAS PubMed; (h) B. Westermann, M. Ayaz and S. S. van Berkel, Angew. Chem., 2010, 122, 858 ( Angew. Chem., Int. Ed. , 2010 , 49 , 846 ) CrossRef; (i) N. T. Jui, E. C. Y. Lee and D. W. C. MacMillan, J. Am. Chem. Soc., 2010, 132, 10015 CrossRef CAS PubMed; (j) Ł. Albrecht, H. Jiang, G. Dickmeiss, B. Gschwend, S. G. Hansen and K. A. Jørgensen, J. Am. Chem. Soc., 2010, 132, 9188 CrossRef PubMed; (k) K. L. Jensen, P. T. Franke, C. Arróniz, S. Kobbelgaard and K. A. Jørgensen, Chem.–Eur. J., 2010, 16, 1750 CrossRef CAS PubMed; (l) T. Urushima, D. Sakamoto, H. Ishikawa and Y. Hayashi, Org. Lett., 2010, 12, 4588 CrossRef CAS PubMed; (m) K. Weiß, S.-W. Wei and S. B. Tsogoeva, Org. Biomol. Chem., 2011, 9, 3457 RSC; (n) S.-W. Wei, K. M. Weiß and S. B. Tsogoeva, Synthesis, 2012, 44, 3441 CrossRef CAS PubMed; (o) M. M. Ibrahim, D. Grau, F. Hampel and S. B. Tsogoeva, Eur. J. Org. Chem., 2014, 1401 CrossRef CAS.
- (a) D. Enders, V. Terteryan and J. Paleček, Synthesis, 2008, 14, 2278 CrossRef PubMed; (b) H. Ishikawa, T. Suzuki and Y. Hayashi, Angew. Chem., 2009, 121, 1330 ( Angew. Chem., Int. Ed. , 2009 , 48 , 1304 ) CrossRef; (c) H. Ishikawa, T. Suzuki, H. Orita, T. Uchimaru and Y. Hayashi, Chem.–Eur. J., 2010, 16, 12616 CrossRef CAS PubMed; (d) H. Ishikawa, M. Honma and Y. Hayashi, Angew. Chem., Int. Ed., 2011, 50, 2824 CrossRef CAS PubMed; (e) G. Dickmeiss, K. L. Jensen, D. Worgull, P. T. Franke and K. A. Jørgensen, Angew. Chem., Int. Ed., 2011, 50, 1580 CrossRef CAS PubMed.
- (a) R. T. Brittain, D. Jack and A. C. Ritchie, Adv. Drug Res., 1970, 197 CAS; (b) D. J. Ager, I. Prakash and D. R. Schaad, Chem. Rev., 1996, 96, 835 CrossRef CAS PubMed; (c) S. C. Bergmeier, Tetrahedron, 2000, 56, 2561 CrossRef CAS and references therein.
- A. Solladié-Cavallo and A. Diep-Vohuule, J. Org. Chem., 1995, 60, 3494 CrossRef.
- S.-W. Wei, R. Messerer and S. B. Tsogoeva, Chem.–Eur. J., 2011, 17, 14380 CrossRef CAS PubMed.
- M. Dukes and L. H. Smith, J. Med. Chem., 1971, 14, 326 CrossRef CAS and references therein.
- L. Almirante and W. Murmann, J. Med. Chem., 1966, 9, 650 CrossRef CAS.
- W. E. Glover, A. D. M. Greenfield and R. G. Shanks, Br. J. Pharmacol. Chemother., 1962, 19, 235 CrossRef CAS PubMed.
- A. Klleman, J. Engel, B. Kutscher and D. Reichert, Pharmaceutical Substances: Syntheses, Patents, Applications, New York, 3rd edn, 1999 Search PubMed.
- Y.-Q. Wen, R. Hertzberg, I. Gonzalez and C. Moberg, Chem.–Eur. J., 2014, 20, 3806 CrossRef CAS PubMed.
- (a) Catalytic Asymmetric Synthesis, ed. I. Ojima, Wiley, New York, 2nd edn, 2000 Search PubMed; (b) J. Marco-Contelles, M. T. Molina and S. Anjum, Chem. Rev., 2004, 104, 2857 CrossRef CAS PubMed; (c) Modern Oxidation Methods, ed. J. E. Bäckvall, Wiley-VCH, Weinheim, 2004 Search PubMed; (d) Q.-H. Xia, H.-Q. Ge, C.-P. Ye, Z.-M. Liu and K.-X. Su, Chem. Rev., 2005, 105, 1603 CrossRef CAS PubMed; (e) For a review on organocatalytic epoxidations, see: K. Weiß and S. B. Tsogoeva, Chem. Rec., 2011, 11, 18 CrossRef PubMed.
- (a) S. T. Oyama, Mechanisms in Homogeneous and Heterogeneous Epoxidation Catalysts, Elsevier, Amsterdam, 2008 Search PubMed; (b) B. Marsh and D. R. Carbery, Annu. Rep. Prog. Chem., Sect. B: Org. Chem., 2008, 104, 35 RSC; (c) D. Díez, M. B. Múñez, A. B. Antón, P. García, R. F. Moro, N. M. Garrido, I. S. Marcos, P. Basabe and J. G. Urones, Curr. Org. Synth., 2008, 5, 186 CrossRef; (d) R. Chawla, A. K. Singh and L. D. S. Yadav, RSC Adv., 2013, 3, 11385 RSC.
- See for example: (a) Y. Tu, Z.-X. Wang and Y. Shi, J. Am. Chem. Soc., 1996, 118, 9806 CrossRef CAS; (b) Z.-X. Wang, Y. Tu, M. Frohn and Y. Shi, J. Org. Chem., 1997, 62, 2328 CrossRef CAS; (c) M. Frohn and Y. Shi, J. Org. Chem., 1997, 62, 2328 CrossRef; (d) Z.-X. Wang and Y. Shi, J. Org. Chem., 1997, 62, 8622 CrossRef CAS; (e) Z.-X. Wang and Y. Shi, J. Org. Chem., 1998, 63, 3099 CrossRef CAS; (f) Y. Zhu, Y. Tu, H. Yu and Y. Shi, Tetrahedron Lett., 1998, 39, 7819 CrossRef CAS; (g) J. D. Warren and Y. Shi, J. Org. Chem., 1999, 64, 7675 CrossRef CAS; (h) H. Tian, X. She, L. Shu, H. Yu and Y. Shi, J. Am. Chem. Soc., 2000, 122, 2435 CrossRef; (i) H. Tian, X. She, H. Yu, L. Shu and Y. Shi, J. Org. Chem., 2002, 67, 2435 CrossRef CAS PubMed; (j) H. Tian, X. She, J. Xu and Y. Shi, Org. Lett., 2001, 3, 1929 CrossRef CAS PubMed; (k) G. A. Cao, Z.-X. Wang, Y. Tu and Y. Shi, Tetrahedron Lett., 1998, 39, 4425 CrossRef CAS; (l) M. Frohn, X. Zhou, J.-R. Zhang, Y. Tang and Y. Shi, J. Am. Chem. Soc., 1999, 121, 7718 CrossRef CAS; (m) L. Shu and Y. Shi, Tetrahedron Lett., 1999, 40, 8721 CrossRef CAS; (n) L. Shu and Y. Shi, J. Org. Chem., 2000, 65, 8807 CrossRef CAS; (o) X.-Y. Wu, X. She and Y. Shi, J. Am. Chem. Soc., 2002, 124, 8792 CrossRef CAS PubMed.
- (a) D. Goeddel, L. Shu, Y. Yuan, O. A. Wong, B. Wang and Y. Shi, J. Org. Chem., 2006, 71, 1715 CrossRef CAS PubMed.
- See for example: http://img1.guidechem.com/msdspdf/110-71-4.pdf.
- C. P. Burke, L. Shu and Y. Shi, J. Org. Chem., 2007, 72, 6320 CrossRef CAS PubMed.
- ESI.†.
- D. Xiong, X. Hu, S. Wang, C.-X. Miao, C. Xia and W. Sun, Eur. J. Org. Chem., 2011, 4289 CrossRef CAS.
- S. Pedragosa-Moreau, C. Morisseau, J. Zylber, A. Archelas, J. Baratti and R. Furstoss, J. Org. Chem., 1996, 61, 7402 CrossRef CAS PubMed.
- B. T. Cho, W. Ki Yang and O. K. Choi, J. Chem. Soc., Perkin Trans. 1, 2001, 1204 RSC.
- B. T. Cho, S. K. Kang and W. K. Yang, Bull. Korean Chem. Soc., 2002, 1328 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 994083. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4ra04011b |
| This journal is © The Royal Society of Chemistry 2014 |