The photocatalytic activity and degradation mechanism of methylene blue over copper(II) tetra(4-carboxyphenyl) porphyrin sensitized TiO2 under visible light irradiation
Huigang Wang*abc,
Dongmei Zhoua,
Shaosong Shena,
Junmin Wana,
Xuming Zheng*a,
Lihong Yub and
David Lee Phillipsb
aDepartment of Chemistry and Engineering Research Center for Eco-dyeing and Finishing of Textiles, MOE, Zhejiang Sci-Tech University, Hangzhou 310018, China. E-mail: zdwhg@163.com; Fax: +86-571-8684-3627; Tel: +86-571-8684-3627
bDepartment of Chemistry, The University of Hong Kong, Pokfulam Road, Hong Kong S.A.R., China
cDepartment of Physics, University of Osnabrueck, 49069 Osnabrueck, Germany. E-mail: huigwang@uni-osnabrueck.de
First published on 19th June 2014
Abstract
Copper(II) meso-tetra(4-carboxyphenyl)porphine (CuTCPP) sensitized nano TiO2 particles, denoted as CuTCPP-TiO2, were synthesized to modify the photoresponse properties of TiO2 particles. Femtosecond and nanosecond spectroscopic studies of CuTCPP and CuTCPP-TiO2 are reported, combining both the time-correlated single-photon counting (TCSPC) technique and transient absorption measurements. The purpose is to investigate the TiO2 photodegradation mechanism and the relationship with the initial electronic and vibrational relaxation of the S1 and S2 excited states of the sensitizer. Excitation of the Soret band leads to multiple electronic and vibrational relaxation time scales of S2 and S1 populations, from hundreds of femtoseconds to tens of nanoseconds. The systematic and detailed studies reported here reveal that the Soret fluorescence band decays with a lifetime of 98–120 ps for CuTCPP whereas 123–398 ps for CuTCPP-TiO2, with the lifetime prolonged, the fluorescence intensities however, are greatly reduced. In addition, the S1 fluorescence was totally quenched after CuTCPP sensitized on the TiO2. The overall picture of electronic relaxation dynamics is depicted according to these observations. The photodegradation of methylene blue (MB) over CuTCPP-TiO2 was systematically investigated. The presence of CuTCPP molecules in the TiO2 surface led to enhancement in its photodegradation of MB under visible light irradiation. The photocatalytic degradation kinetics of MB fitted well with the Langmuir–Hinshelwood mode. Moreover the detailed short-time dynamics for the visible-light induced catalytic mechanism was discussed.
1. Introduction
Most of the developed and developing industrialized nations are faced with a tremendous set of environmental problems related to the remediation of hazardous wastes and contaminated groundwater.1–3 The industrial waste materials are very complex; presently physical techniques are the key treatment methods including adsorption,4–6 sedimentation,7 filtration8 as well as flocculation.7 Although these methods can transfer the pollutants from aqueous solution to solid phase, the total quantity or hazardous potential of the pollutants are recalcitrant in nature. To destroy organic pollutants in water one of the most important processes is heterogeneous photocatalysis via semiconductor.9,10 The vital draw-back of TiO2 or ZnO semiconductors is that they absorb only the ultraviolet radiation whose wavelength is less than 387 nm (λ ≦ 385 nm), which only occupy less than 5% of whole sunlight.11,12 Dye sensitization is considered to be a viable alternative method to modify the photoresponse properties.11,13–15 Over the last 30 years the scientific and engineering interest in the application of dye sensitized semiconductor photocatalysis has grown exponentially.13–15Porphyrins are recognized to be the most promising sensitizers. In particular, metal porphyrin have been shown to be efficient photosensitizers and catalysts for a variety of oxidation reactions.16–20 Recently, the photocatalytic activity of TiO2 powders impregnated with Cu porphyrins has been investigated and shown to be the best one on the decomposition of 4-nitrophenol (4-NP).18,20
In addition to the extraordinary experimental achievements, some mechanistic aspects and time resolved techniques have been applied to investigate on the catalytic mechanism and electronic transfer dynamics.17 Spectroscopic investigations shown that the relaxation dynamics of porphyrins is significantly modulated by the central metal ion in metalloporphyrins and the substitution in the macrocycle.21–23 On the basis of laser flash photolysis measurements general mechanism as well as the corresponding primary reaction rate for heterogeneous photocatalysis on TiO2 have been proposed.24 Griffith etc. investigated systematically the influence of porphyrin molecule structure on efficiency determining electron transfer kinetics and solar cells performance. But the research was limited in free-base and zinc porphyrin dyes.17
After metal ions quantitatively coordinate in the central cavity, the molar extinction coefficient of porphyrin will be strongly enhanced and the photoresponse in the visible light region is extended as well. Cu(II) porphyrins (CuTCPP) are usually four coordinated with ligands and stable against the acid-induced demetallation. With the partially filled d orbitals, Cu(II) ions are capable of fluorescence quenching by electron or energy transfer.25,26 Furthermore, Cu(II), with its d9 valence electron configuration, is paramagnetic, which has be shown to increase the quenching efficiency of the metal ion.25,26 Therefore, the excited state of Cu(II) porphyrin (CuTCPP*) has a high possibility for electrons to inject to the conduct band of TiO2, which is exactly different to the behavior of well-known zinc(II) porphyrins.
Moreover, Cu(II) porphyrin belong to the different group of zinc(II) porphyrins in Metalloporphyrins based on their spectroscopic properties.27 Regular metalloporphyrins contain closed-shell metal ions (d0 or d10)—for example Zn2+, in which the dπ metal-based orbitals are relatively low in energy. These have very little effect on the porphyrin π to π* energy gap in porphyrin electronic spectra. Hypsoporphyrins are metalloporphyrins in which the metals are of dm, m = 6–9, having filled dπ orbitals—for example Cu2+. In hypsoporphyrins there is significant metal dπ to porphyrin π* orbital interaction (metal to ligand π- backbonding).27 This results in very different photoexcited electronic reactions.
Based on our previously reported works on the Soret(Bx and By-band) electronic transitions and the corresponding photo relaxation dynamics of tetra (4-carboxyphenyl) porphyrin (TCPP),28,29 and the work on degradation mechanism of methyl orange under photo-catalysis of TiO2,30 in this work, copper(II) meso-tetra(4-carboxyphenyl)porphine sensitized TiO2 (CuTCPP-TiO2) were synthesized and their properties of photo-generated reactive species, holes and electrons were studied by transient absorption spectroscopies and time-correlated single-photon counting (TCSPC) technique. Photocatalysis of methylene blue (MB) over CuTCPP-TiO2 were systematically investigated.
2. Experimental methods
2.1 Materials
Meso-tetra(4-carboxyphenyl) porphyrin were purchased from J&KCHEMICA with no further purification. TiO2 nanoparticles (P25, d = 21 nm) were purchased from Beijing Entrepreneur Science & Trading Co. DMF and MB was of analytical reagent grade quality and was used without further purification. Other chemicals were commercial products of analytical grade or reagent-grade. All the solutions were prepared with distilled water.2.2 Synthesis
A 27.0 mg (0.15 mmol) of CuCl2 were added to 42.4 mg (0.05 mmol) of the TCPP dissolved in 100 mL of DMF. The mixture was heated to reflux for 8 h and monitored by TLC until the complete disappearance of the starting material TCPP. The unreacted solid salt was filtered off and the solvent was removed under vacuum. The crude product was washed with water and then purified by chromatography on a silica gel column with CH3OH![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :CHCl3 = 1:3 as eluant. The product CuTCPP was recovered in nearly quantitative yield. 1H NMR(CDCl3, 400 MHz): δ, ppm 8.9 (s, 8H), 8.4 (d, 8H), 8.3 (s, 8H). UV-vis (CHCl3): λmax, nm, 418 (Soret band), 540 (Q bands).
:CHCl3 = 1:3 as eluant. The product CuTCPP was recovered in nearly quantitative yield. 1H NMR(CDCl3, 400 MHz): δ, ppm 8.9 (s, 8H), 8.4 (d, 8H), 8.3 (s, 8H). UV-vis (CHCl3): λmax, nm, 418 (Soret band), 540 (Q bands).
The TiO2 sensitized by CuTCPP were obtained as follows: 1 g TiO2 was added into the 100 mL 0.5 mmol L−1 DMF solution of CuTCPP. The mixture was refluxed for 5 h in dark, and then filtration to get the solids, washed with 10 mL DMF 5 times and then washed with distilled water 5 times, the solids were dry, in this way, TiO2 coupled by CuTCPP were prepared, and denoted as CuTCPP-TiO2.
2.3 Characterization
Transient Fluorescence Decay measurements were performed by using the Time Correlated Single-Photon Counting (TCSPC) technique with a confocal fluorescence microscope system (HORIBA, Ltd.). The excitation light source was a mode-locked Ar + ion laser centered at 370 nm, operated at a frequency of 8 MHz. The fluorescence photon was collected between 540 nm and 580 nm and no counts of fluorescence from the TiO2 or quartz substrate was detected. The femtosecond TA experiment was carried out by a femtosecond Ti:Sapphire regenerative amplified Ti:Sapphire laser system (Spectra Physics, Spitfire-Pro) and an automated data acquisition system (Ultrafast Systems, Helios). The pump laser was of 267 nm (the third harmonic of the fundamental 800 nm from the regenerative amplifier). The probe pulse was white-light continuum (450–800 nm) generated by a sapphire plate using approximately 5% of the original output from the Spitfire. Before passing through the sample the probe pulse was split into two beams: one beam travels through the sample, and the other is sent directly to a reference spectrometer that monitors the fluctuation of the probe beam intensity. The instrument response function is evaluated to be 150 fs. The nanosecond TA experiment was carried out using a LP920 laser flash spectrometer manufactured by Edinburgh Instruments Ltd. The probe light source was a 450 W ozone free Xe arc lamp producing a continuous spectrum between 150 and 2600 nm. The pump light was generated by a Q-switched Nd:YAG laser (Model: Lab 150-10 4056L; 3rd harmonic line at l = 355 nm). The TA signal was recorded with a TDS 3012C digital signal analyzer. The TA signals of all samples in the nanosecond TA experiment with the occurrence of the photocatalytic reactions were reproducible, and therefore the accumulation of photo-catalytic products was negligible.The timedependent TA signals at different pump and probe wavelengths were fit to a multiexponential function, convoluted with a Gaussian instrument response function of 120 fs FWHM. The multiexponential function, (f(t)) = 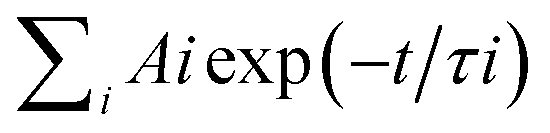 commonly used in TA data analysis, was derived on the assumption of first order reaction kinetics with parallel pathways from the first excited state.
commonly used in TA data analysis, was derived on the assumption of first order reaction kinetics with parallel pathways from the first excited state.
The degradation rates of MB solutions were periodically scanned by a UV-2501 spectrophotometer (Shimadzu, Japan), and the maximum absorption wavelength of MB solution was identified at 665 nm. UV-vis spectra data were recorded in the range from 200 to 800 nm. The X-ray diffraction (XRD) patterns were recorded on D5000 Diffraction system. XPS measurements were performed with Perkin Elmer PHI 5900 and equipped with non monochromatized source (operating at 150 W).
2.4 Photocatalytic activity measurements
The photocatalytic activity of CuTCPP-TiO2 samples was evaluated by the degradation rate of methylene blue under visible-light irradiation. The experimental set up placed in a black box consisted of a 200 mL beaker irradiated by a 250 W, 220 V iodine-tungsten lamp. The distance between the lamp and the solution is 50 cm. A 420 nm cutoff filter (Schott Glass) was placed between the lamp and the beaker in order to absorb the UV light. The temperature inside the reactor was maintained at ca. 298 K by means of a continuous circulation of water surrounding the reactor.100 mL methylene blue (MB) aqueous solution (0.9 mg L−1) was mixed with 20.0 mg photocatalyst powder in the above 200 mL beaker. The mixture was stirred with a magnetic bar and bubbled with oxygen. Keep the mixture 50 min in darkness before switching on the lamp to allow the physical adsorption reach equilibrium. The initial pH value of the suspension was 4.0 and it was measured and keeps in a narrow range during the photocatalytic experiments.
The photoreactivity runs lasted 400 min. Samples of 3 mL were withdrawn from the suspension every 50 min during the irradiation. The photocatalysts were separated from the solution by centrifugation and the quantitative determination of methylene blue was performed by measuring its absorption at 664 nm with a Shimadzu UV-2501 PC spectrophotometer. For comparison, the MB photodegradation experiment was conducted under the same conditions using Degussa P25 powder as photocatalysts.
3. Results
Understanding the dynamic processes observed in the time resolved spectra preliminary requires a deep insight into the static spectroscopic properties of the sample. Here we presented the absorption and fluorescence spectra of CuTCPP in tetrahydrofuran (THF) in Fig. 1, the fluorescence spectra of CuTCPP-TiO2 is also included. Fig. 1 shows that, the Soret band and the Q band. The strongest absorption Soret band at 417.6 nm is assigned as B (0,0), where the numbers in parentheses indicate the number of quanta of the dominant Franck–Condon active vibrational mode in the upper and lower electronic states of the transition, respectively. The Q band is at 540.4 nm and assigned as Q(0,0). The lowest triplet state, T1, lies below the Q(0,0) state.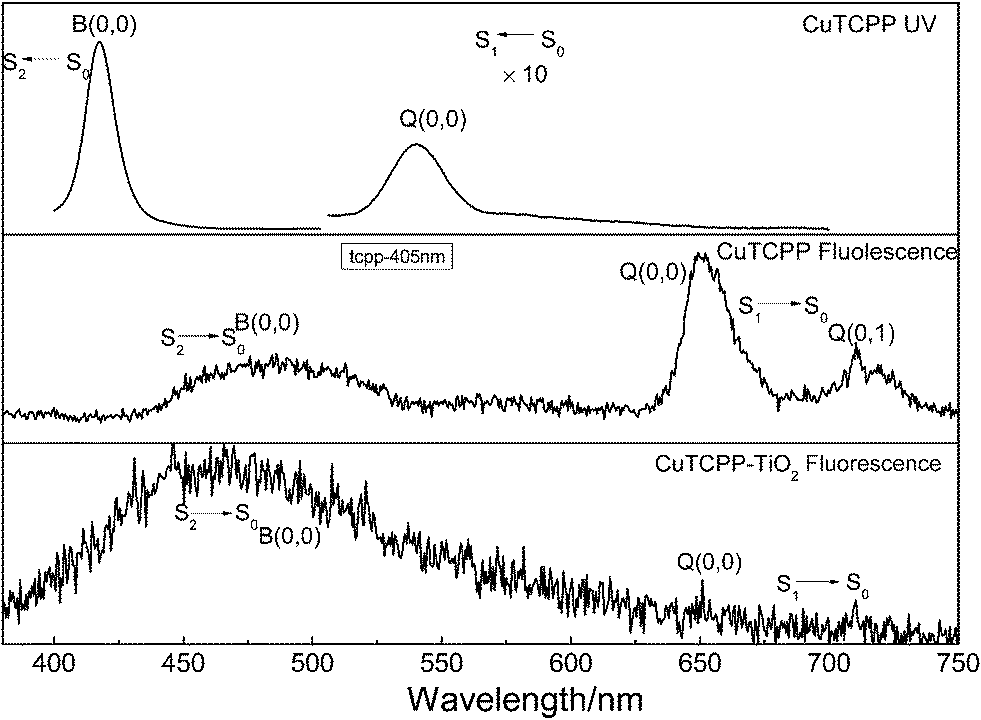 | ||
| Fig. 1 The UV-vis absorption and static fluorescence spectra of CuTCPP in tetrahydrofuran solution and the fluorescence spectra of CuTCPP-TiO2 in solid. For the fluorescence spectrum, the excitation wavelength was 365 nm, the sample concentration was 1 μM in a 5 mm cell, reabsorption may be significant below 430 nm. | ||
The CuTCPP and CuTCPP-TiO2 fluorescence spectra in Fig. 1 for 365 nm excitation clearly show S2 → S0 fluorescence. Peaks in S1 → S0 fluorescence of CuTCPP are measured at 651.5 nm for Q(0,0) and 711.5 nm for Q(0,1). Correspondingly the S1 → S0 fluorescence of CuTCPP-TiO2, however disappeared, the emission intensity of Q(0,0) were quenched by TiO2. The peak in S2 fluorescence of CuTCPP is measured at 487.7 nm, whereas of CuTCPP-TiO2 is measured at 469.0 nm, blue-shifted 18.7 nm after anchored to TiO2, and the intensity is weakened. The interaction of the carboxylate group with surface Ti ions is likely to lead to the formation of Ti–O–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds. The carboxylate serves, therefore, as an interlocking group enhancing electronic coupling between the π orbitals of the phenyl and the Ti(3d) orbital manifold of the semiconductor. This coupling is rendered efficient by the presence of π electrons in the bridging group leading to increased delocalization of the π level of the phenyl and pyrrole ligand. The energy of the π level is decreased by this delocalization, thus increased the energy gap between S0 and S2 state, which explains the observed blue shift in the fluorescence spectrum.
O bonds. The carboxylate serves, therefore, as an interlocking group enhancing electronic coupling between the π orbitals of the phenyl and the Ti(3d) orbital manifold of the semiconductor. This coupling is rendered efficient by the presence of π electrons in the bridging group leading to increased delocalization of the π level of the phenyl and pyrrole ligand. The energy of the π level is decreased by this delocalization, thus increased the energy gap between S0 and S2 state, which explains the observed blue shift in the fluorescence spectrum.
The only pathway for electrons in S1 excited state in CuTCPP comes out to be 3 possible relaxations pathways after CuTCPP anchored to TiO2-transferred to the TiO2 conduction band, transferred to the triplet state or transferred to S0 state, and consequently quenched the emission intensity of fluorescence S1 → S0 process.
Fig. 2 shows the fluorescence decay profiles of CuTCPP (solid) and CuTCPP-TiO2 (solid) respectively. The fluorescence intensity of CuTCPP-TiO2 have been multiplied by a factor of 1.54, this means that in our experiment, the 440 nm fluorescence of CuTCPP is a bit quenched by TiO2 after UV excitation (370 nm), and its lifetime becomes longer as shown in Fig. 2 (fitted with exponential function to get the lifetime of 98.2 ps for CuTCPP and 123.1 ps for CuTCPP-TiO2). In a typical electron transfer process from a dye (for example N719) to anatase TiO2 under UV excitation, the fluorescence intensity of the dye is usually quenched meanwhile with its lifetime shortened to some extent.31 The longer life time keeps the electron–hole separation longer enough to take place the following photochemical reactions.
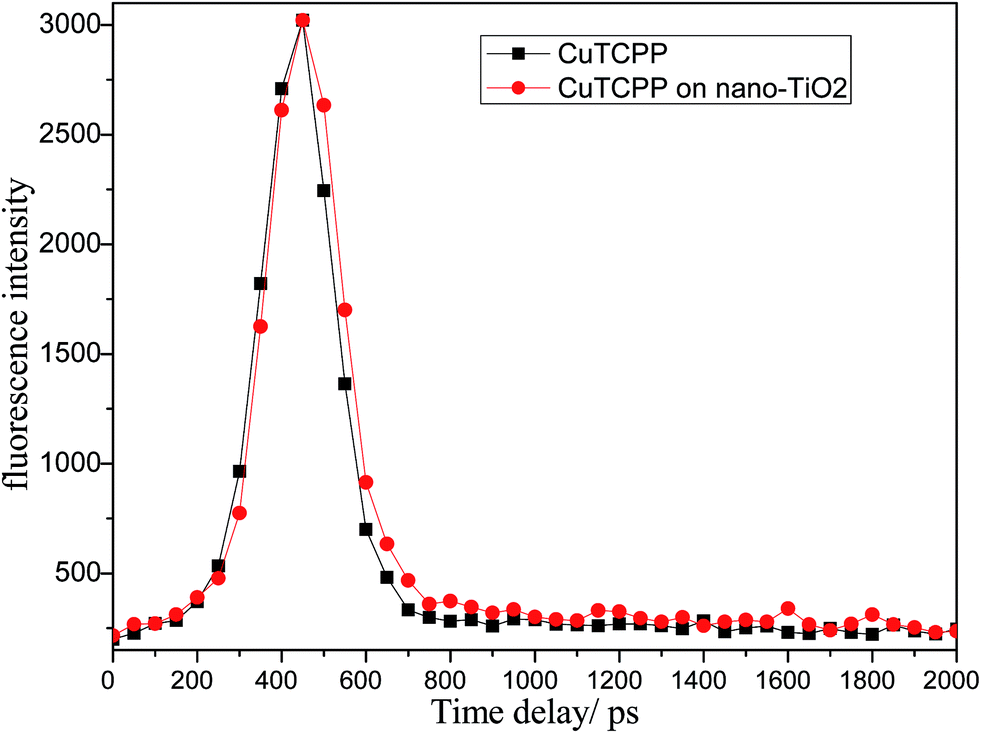 | ||
| Fig. 2 The normalization of the fluorescence decay profiles of CuTCPP and CuTCPP-TiO2 (multiplied by 1.54) obtained by TCSPC. | ||
It is well known that the recombination of photo-generated electrons and holes in nano-sized TiO2 occurs in several microseconds under vacuum conditions. We also characterized the CuTCPP-TiO2 with Transient absorption spectroscopy in the femto and nanoseconds to study the recombination processes of electrons and holes.
Fig. 3 presents the femtosecond (A) and nanosecond (B) TA spectra of CuTCPP and CuTCPP-TiO2 dispersed in MeCN. We used MeCN as the solvent to disperse the CuTCPP and CuTCPP-TiO2 because acetonitrile (MeCN) is a slow holes scavenger of TiO2 and the reaction between MeCN and holes is not detected on the femtosecond time scale. The TA spectra were obtained by subtracting the absorption spectrum in the original state from the spectrum measured at certain delay time. After Soret band wavelength photo excitation, three distinct bands could be clearly observed in CuTCPP in femtosecond TA(Fig. 3A), one strong band sited at 491.5 nm and other two broad bands with peaks sited at 567.2 nm and 594.3 nm respectively. While only two weak bands could be detected in CuTCPP-TiO2 in femtosecond TA(Fig. 3A), one band at 522.2 nm and another broad band sited at 634.5 nm, as for nanosecond TA(Fig. 3B), the 390.3 nm TA band in CuTCPP was blue shift to 383.5 nm in CuTCPP-TiO2. The negative bands at 450 nm (assigned as fluorescence of the CuTCPP) in CuTCPP disappeared in CuTCPP-TiO2 and additionally there seems a very broad band across the whole region from 450 to 750. This broad absorption band comes from the electron–holes separation states of TiO2 nanoparticles. That is, the absorptions of holes and electrons overlapped with each other in the broad absorption range. According to the literature, the band centered at 520 nm and 1200 nm are assigned as trapped holes on TiO2 and the band centered at 770 nm is assigned as trapped electrons.32 A precise assignment of the absorptions of electrons and holes is rather difficult because their absorption spectra are very broad and the bands overlap each other. In Fig. 3B, the very broad absorption band range from 450 nm to 750 nm suggest that some holes and electrons are photogenerated and are trapped separately.
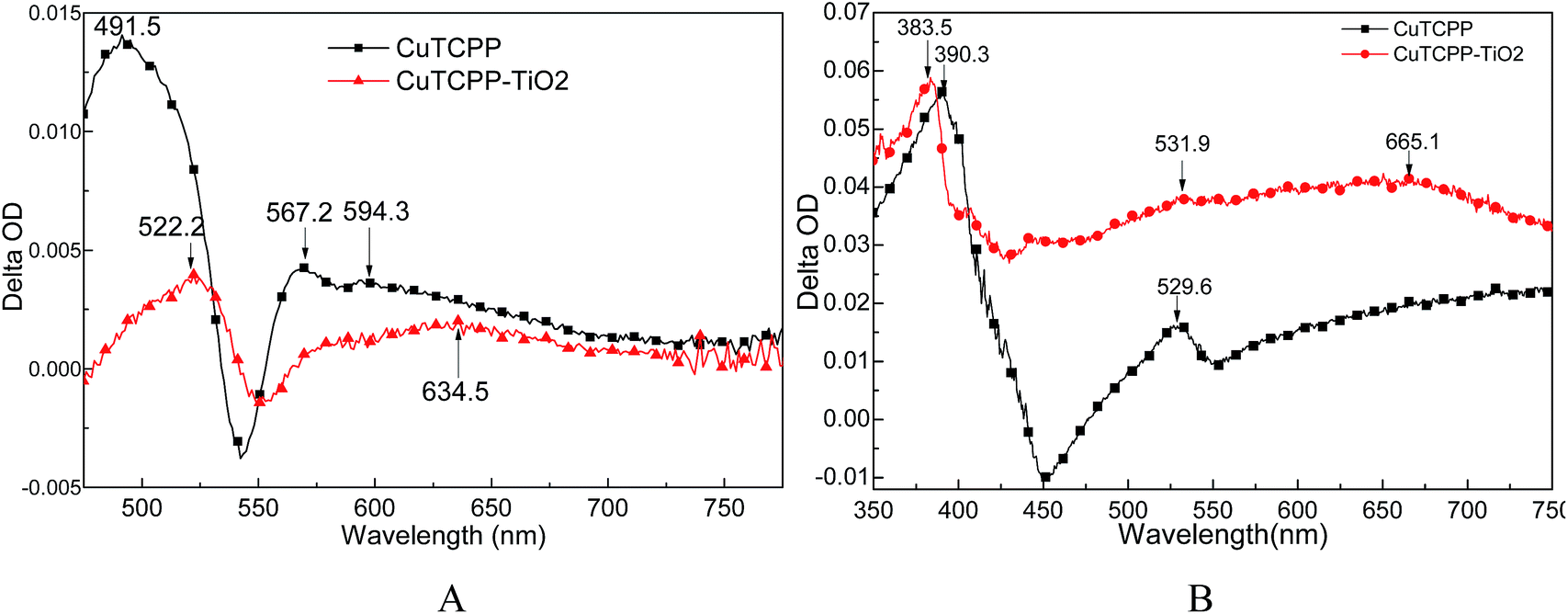 | ||
| Fig. 3 Transient absorption spectra of CuTCPP and CuTCPP-TiO2: (A) collected 2 ps delay after exciting at 400 mm laser in the spectral range 475–775 nm with femtosecond pulses. (B) Collected 1 ns delay after exciting at 355 mm laser in the spectral range 350–750 nm with nanosecond pulses. | ||
Since the transient absorption patterns shows very different after CuTCPP anchored to TiO2 nanoparticles as shown in Fig. 3, undoubtedly the kinetic behaviors shows different way also as discussed below.
Fig. 4 shows the femtosecond transient decay profile of CuTCPP measured at three different probe wavelengths when pumping the Soret band. The transient at 491.5 nm rises to the maximum at ∼1.21 ps and then decays on the picosecond time scale. The transients at 567.2 nm and 594.3 nm rise to the maximum at ∼3.30 ps simultaneously and then decay individually. The maximum of transient at 567.2 nm and 594.3 nm are lag 2.09 ps behind that of transient at 491.5 nm. The TA at 491.5 nm shown in Fig. 4 was well fit with three picosecond exponential components (lifetimes 0.90, 19 and 120 ps). As to the relaxation dynamics of the transient at 567.2 nm and 594.3 nm, note that they appears as a rise after the inflexion (1.21 ps) by the decay of the transient at 491.5 nm, The fit parameters are given in Table 1, the nanosecond TA fit parameters are also included. Three different femtosecond range lifetime and one nanosecond lifetime components contribute to the signal of the relaxation dynamics of CuTCPP. Note that the femtosecond lifetime appears as a rise at 567.2 nm and 594.3 nm while appears as decline at 491.5 nm, which consistent with the inter conversion process of S2 → S1. The broad picture confirms general features of the ultrafast dynamics and be represent schematically in Fig. 5, this is similar to previous studies in other porphyrins:21,33 the lifetime of τ3 is in consistency with the characterization of TCSPC in Fig. 2, so we assigned the τ3 as the S2 → S1 fluorescence process. The overview of the dynamic process describes as: For excitation around 400 nm, relaxation of S2 by internal conversion (IC) and the rise of S1 occur in about 0.9 ps, followed by a slower vibrational relaxation by energy loss to the solvent on a time scale on the order of 6–19 ps and S2 → S0 fluorescent process on a time scale of 100–120 ps, and finally S1 decay (intersystem crossing + fluorescence), time constant 16–18 ns.
 | ||
| Fig. 4 Transient absorption decay profile of CuTCPP dispersed in MeCN at three different probe wavelengths. | ||
| λprobe | Lifetimes | Relative amplitudesa | ||||
|---|---|---|---|---|---|---|
| τ1 | τ2 | τ3 | A1 | A2 | A3 | |
| a A negative amplitude indicates that the respective component is a rise instead of a decay. | ||||||
| 491.5 | 0.90 | 19 | 120 | 0.0040 | 0.0117 | 0.0026 |
| 567.2 | 0.55 | 7 | 120 | −0.0091 | 0.0029 | 0.0034 |
| 594.3 | 0.50 | 6 | 101 | −0.0088 | 0.0029 | 0.0020 |
| 390.3 | 17173 | 5.52 × 1030 | ||||
| 529.6 | 16827 | 1.04 × 1031 | ||||
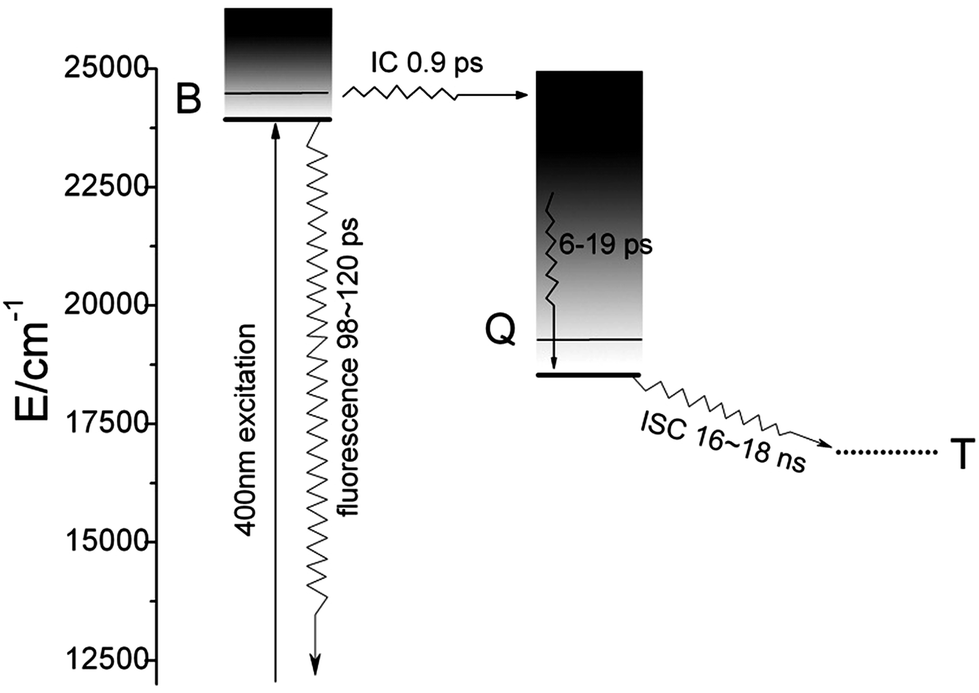 | ||
| Fig. 5 Schematic diagram of the energy relaxation dynamics of CuTCPP. Solid horizontal lines are located at the energies of bands in the absorption spectrum. Dashed lines indicate approximate energies for T1. See text for details of the dynamics. | ||
For the case of CuTCPP-TiO2, the relaxation of S2 process is very similar to CuTCPP, however the every lifetime of components step is different, which have been show in Table 2. Especially, the S2 and S1 state of CuTCPP-TiO2 have been quenched as shown in Fig. 1, so the TA of femtosecond spectra is very weak, and made the fitted lifetime ambiguous, τ3 is largely different from the TCSPC result. the nanosecond spectra however is stronger than CuTCPP, as shown in Fig. 3B. Although the S1 state is completely quenched as shown in Fig. 1, the electron–holes separation states of TiO2 nanoparticles is appears in this absorption region. In general, the relaxation of S2 by internal conversion (IC) and the rise of S1 occur in about 0.5 ps, followed by a slower vibrational relaxation by energy loss to the solvent on a time scale on the order of 24–33 ps and S2 → S0 fluorescent process on a time scale of 123 ps, and finally the electron–holes separation states decay, time constant 36–47 ns.
| λprobe | Lifetimes | Relative amplitudesa | ||||
|---|---|---|---|---|---|---|
| τ1 | τ2 | τ3 | A1 | A2 | A3 | |
| a A negative amplitude indicates that the respective component is a rise instead of a decay. | ||||||
| 491.5 | 0.43 | 24 | 319 | 0.0015 | 0.0012 | 0.0015 |
| 521.0 | 0.43 | 33 | 382 | −0.0092 | 0.0018 | 0.0026 |
| 632.2 | 0.33 | 26 | 398 | −0.0116 | 0.0009 | 0.0014 |
| 383.5.3 | 36578 | 9.98 × 1013 | ||||
| 600 | 46883 | 6.59 × 1010 | ||||
In general, there are two types of charge recombination channels.17 The first is a fast (geminate) recombination process, which occurs between an oxidized dye and the injected electron that originates from the same dye. Matthew JG reported the very interesting sub-ns (0.5 ns to 100 ns) charge recombination channel between photo-injected electrons and porphyrin cations recently, the second process proceeds on a millisecond time scale and occurs between electrons and either oxidized dye molecules or the redox mediator. Our observation is in consistency to the fast recombination process and be assigned as the recombination between oxidized dye and the injected electron.
Phase composition
XRD was carried out to investigate the crystal identity of TiO2 samples and the effect of CuTCPP on the crystal structure of TiO2. Fig. 6 shows the typical XRD pattern of CuTCPP-TiO2 and P25 nanoparticles. The P25 powder contains 20% rutile and 80% anatase. The diffraction peaks at 2θ = 25.3, 37.8, 48.0, 53.1, 55.4, 62.7, 68.8, corresponds to the (101), (004), (200), (105), (211), (204),and (116) planes of anatase TiO2, respectively. The peak of which were strong and in good agreement with the standard XRD patterns of anatase (JCPDS no. 84-1286). While the peak at 27.4°,36.1°and 41.5° are also present, indicating there coexists the rutile phase (JCPDS no. 88-1175). Based on the Scherrer formula, the crystallite sizes of P25 is about 20 nm and the CuTCPP-TiO2 is 20 nm. These results indicate that the CuTCPP sensitized on the surface of TiO2 hardly has any effect on the crystal structure of TiO2 powders.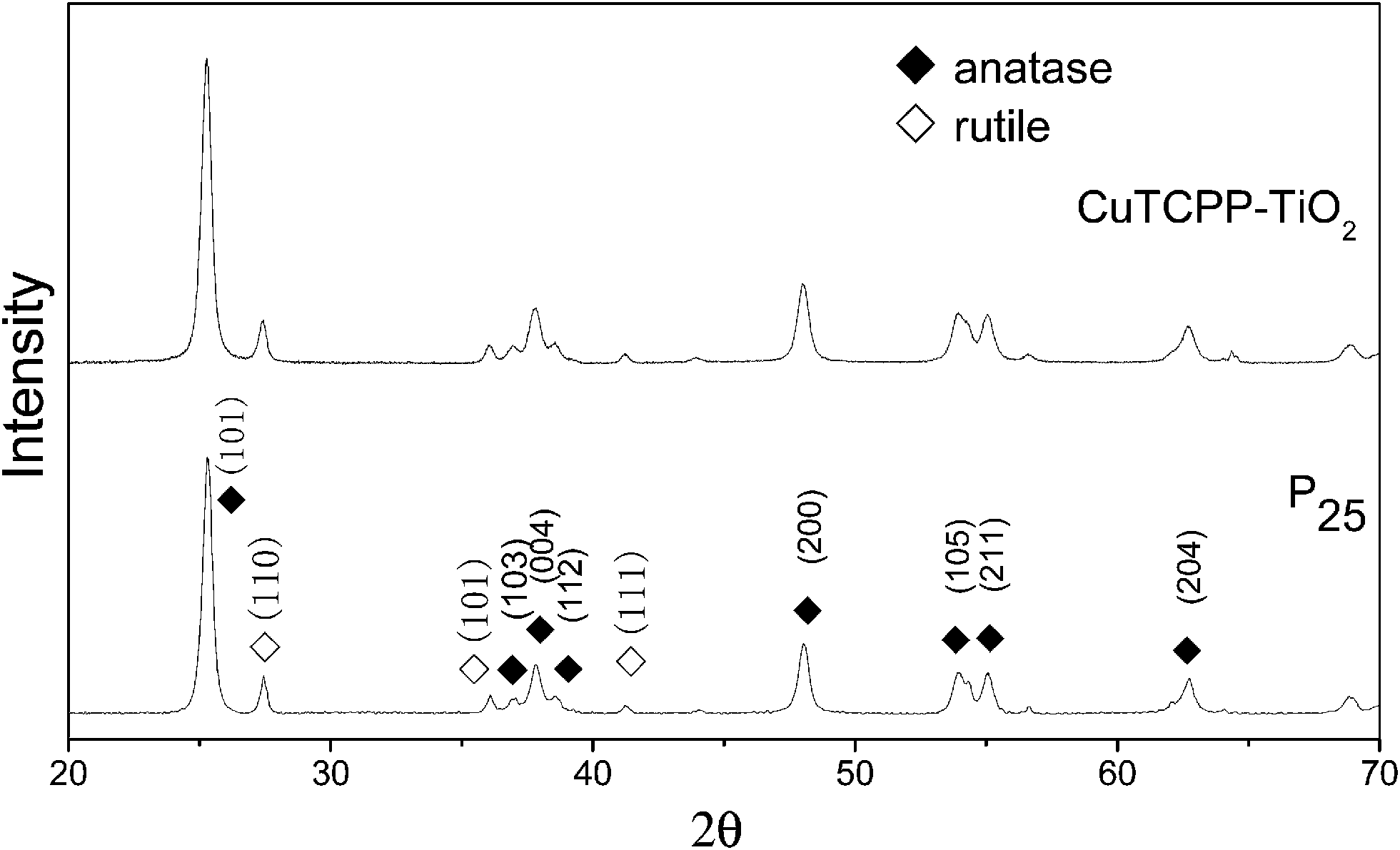 | ||
| Fig. 6 XRD patterns of CuTCPP-TiO2 and P25. ◆ stands for anatase, ◇for rutile. | ||
XPS was used to distinguish the surface change of pure TiO2 and CuTCPP-TiO2. Fig. 7 and 8 show Ti 2p and O 1s XPS spectra of pure TiO2 and CuTCPP sensitized TiO2 respectively. The Ti (2p3/2) binding energy values of TiO2 and CuTCPP-TiO2 are 455.8 and 455.9 respectively. The peak of CuTCPP-TiO2 is higher 0.1 eV than that of pure TiO2. This data suggest that H atom as the donor coordinates with oxygen atom in Ti–O–H in TiO2 and that the oxygen atom accepts electrons. When the CuTCPP sensitized to TiO2 the Carbonyl group substitute the H as Ti–O–CO, contrary to the H group, carbonyl is a electron-accepting groups, it withdraw electrons from O atoms and then raised the valence of Ti atom, decreased the density of the layer electron cloud, leading to the increase in the binding energies of the Ti (2p3/2). The attraction of electron from O atom also leading to the decrease of the electron cloud density of O atom, raising the valence of O atom and consequently increase in the binding energies of the O 1s, which can be demonstrated in O 1s XPS spectra shown in Fig. 7. The adsorption mechanism of CuTCPP on the TiO2 surface can be attributed to the weak chemisorptions.
 | ||
| Fig. 7 XPS of the Ti 2p regions. | ||
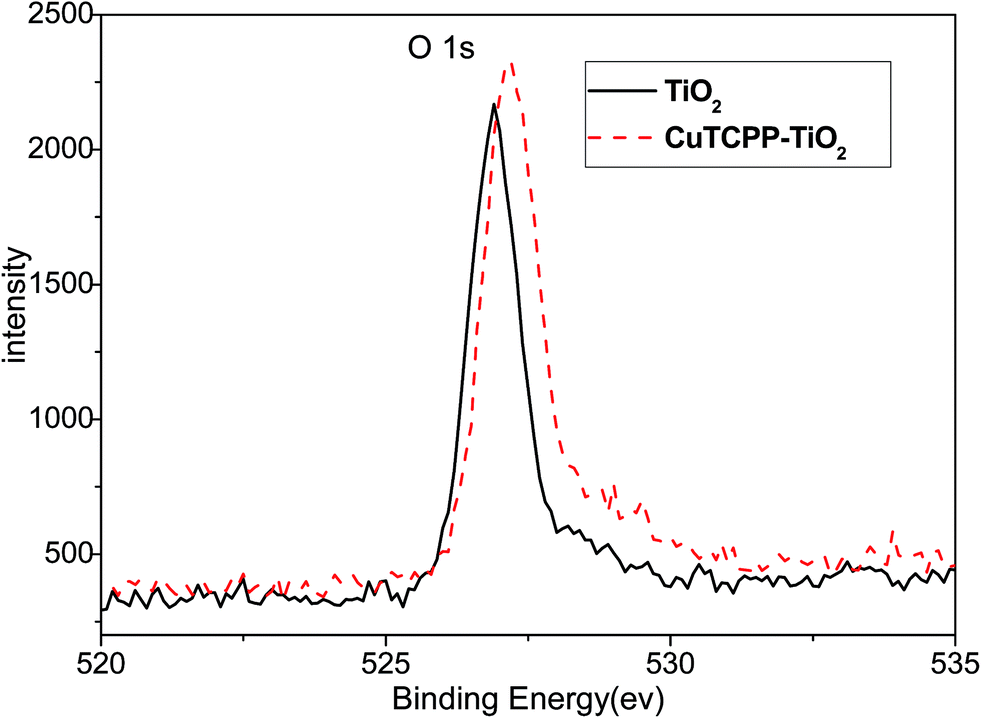 | ||
| Fig. 8 XPS of the O 1s regions. | ||
Photocatalytic activity of CuTCPP-TiO2
It is well known that the photocatalysis activity depends on the crystallinity, phase, and the surface area of the materials. In this work, the visible light induced photocatalytic activities of the as-synthesized CuTCPP sensitized TiO2 were evaluated by photocatalytic degradation of MB under 250 W iodine-tungsten lamp illuminations with 420 nm cutoff filter. Our previous research shows that the adsorption process takes very quickly, completed in the first 50 minutes. Based on this date of absorbance experiments in the dark, all degradation reactions were allowed to stand in the dark for a period of 50 min to establish adsorption equilibrium, and then performs the visible light irradiation. As shown in Fig. 9, The CuTCPP-TiO2 can efficiently photo degrade MB under visible light irradiation. When the initial concentration 9 mg L−1 of MB was used, adding different amounts of CuTCPP-TiO2 (20, 40, 60 and 80 mg) showed 59.28%, 68.76%, 79.47% and 85.02% decolorization rates (1−ct/c0), respectively, under iodine–tungsten lamp illumination after 400 min. Accordingly dark adsorption contributed 38.30%,56.52%, 69.91% and 77.53% to the reduction of MB concentration, respectively, indicating the significant photodegrade capacity of the CuTCPP-TiO2 in the whole process. The high efficiency performance could be ascribed to its unique cooperative effects of strong adsorption ability and low band gap energy.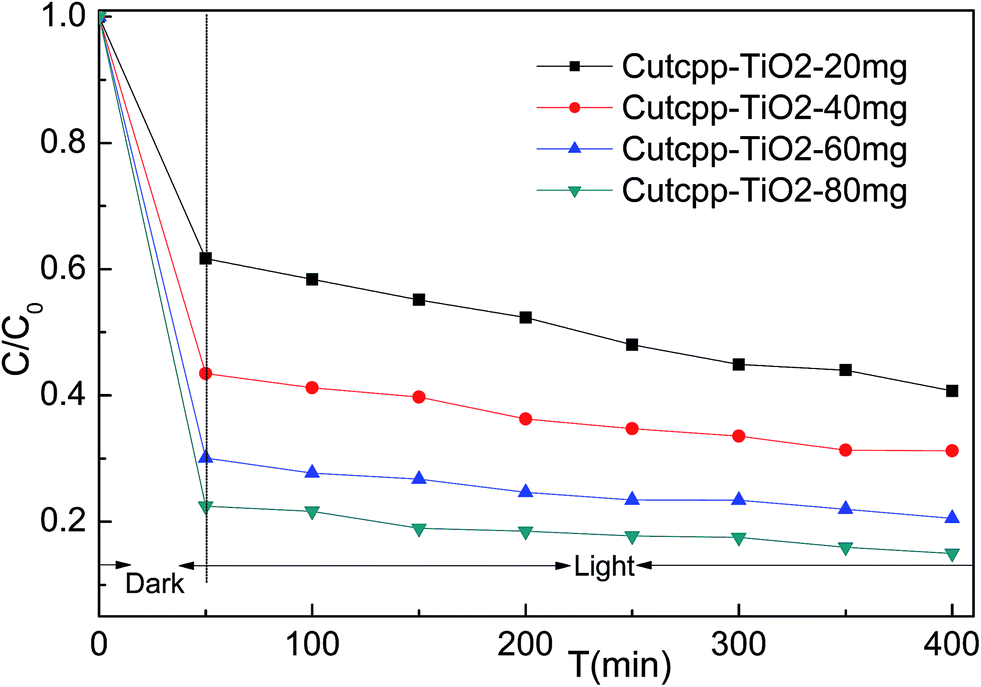 | ||
| Fig. 9 Evolution of MB concentration as a function of time: 50 min in the dark followed of 400 min under visible light irradiation. | ||
During the irradiation period, MB-photo degradation data were analyzed using the Langmuir–Hinshelwood kinetic model:34 with the rate 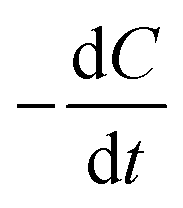 being proportional to the coverage θ which becomes proportional to C at low concentrations:
being proportional to the coverage θ which becomes proportional to C at low concentrations: 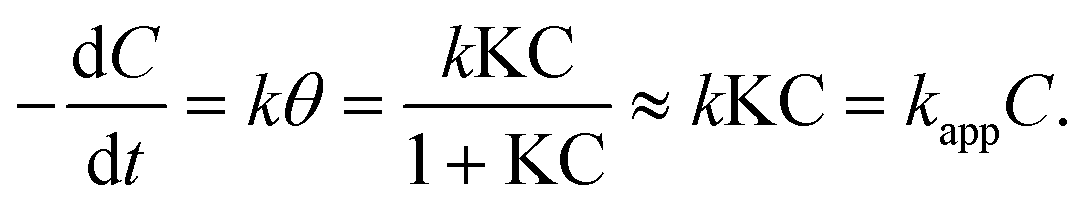
In all cases, a satisfactory linearity was obtained by applying Langmuir–Hinshelwood kinetic model (Fig. 10). Values of the apparent first-order constant, kapp, keeps constantly for different amount of CuTCPP-TiO2, indicating it have no relation with the adding dose of catalysts.
 | ||
| Fig. 10 First order linear fitting of disappearance of MB by photocatalysis under visible light irradiation. | ||
4. Discussion
The first conclusion that we can draw from our data is that the CuTCPP is chemical linked to the anatase surface. Obviously, the only physical absorption between the sensitizer and anatase could not appreciably affect the electron states in each material. Evidences of a chemical Ti–O–CO link are provided by the fact that the blue-shift of the fluorescence spectra of CuTCPP-TiO2 and the obvious binding energies changes in XPS spectra, as shown in Fig. 1–3,7 and 8. The (101) anatase surface is the mostly exposed area for anatase nanocrystals,35 which we are using for the dye sensitizing in photodegradation. It is clear from Fig. 11 that the electron hopping along the Ti–O–CO chain is the main mechanism of the charge transport between the dye and the surface (101) of anatase. The C–O bond connects the sensitizer molecule to anatase, whereas the oxygen atom belongs to both materials.
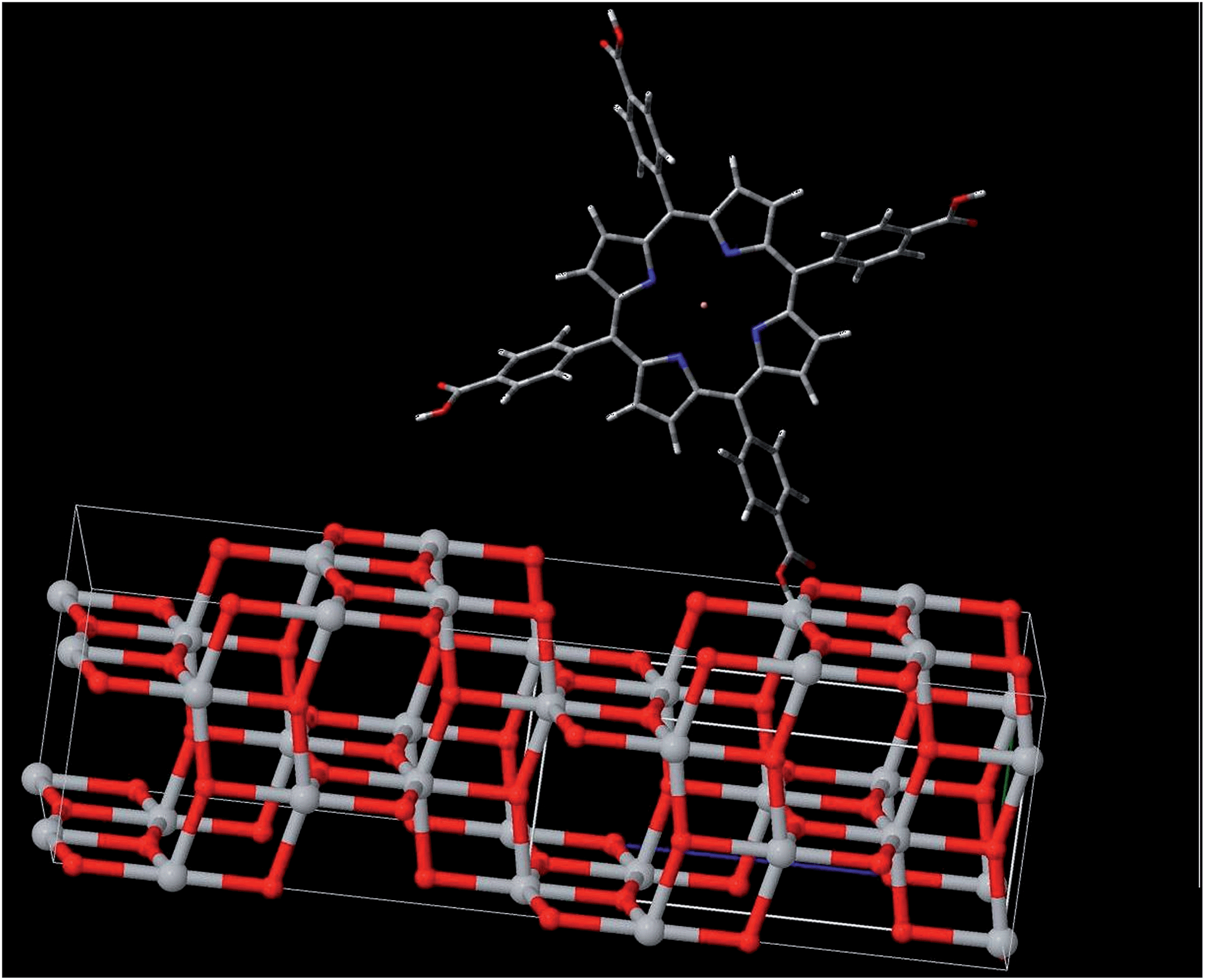 | ||
| Fig. 11 Scheme showing the molecular structure for CuTCPP anchoring to the (101) surface of anatase. | ||
The fluorescence spectra of CuTCPP and CuTCPP-TiO2 demonstrated that, the emission from the S1 state to S0 state in the Franck–Condon active mode in CuTCPP is greatly quenched by the linking of TiO2, and the emission from the S2 state is expected to lie at 487.7 nm, however occurred at 469.0 nm after link to TiO2. These indicate that the coupling of π orbitals of the porphyrin with the Ti(3d) orbital opens the relaxation way of the electron transportation to CB1 state of TiO2, and therefore, quenched the fluorescence intensity in S1–S0 process as observed in Fig. 1. Then to what physical processes in the excited states do the observed lifetimes correspond? The lifetime fitted in Fig. 2 could be definitely assigned as the lifetime of S2 → S0 fluorescence process, then what is the lifetime fitted in nanosecond transient absorption decay profile in Table 2? 680 nm TA absorption is assigned as the shallow trapped conduction-band electron, so here we can assign the TA fitted lifetime as the recombination lifetime of trapped electron (see eqn (4)). And we find that the lifetime hasn't been changed after the TiO2 sensitized with CuTCPP. The lifetime of this electron trapped state is vital for photocatalysis quantum efficiencies.
On the basis of the ref. 24 and our dynamic measurements we proposed the following mechanism for heterogeneous photocatalysis on CuTCPP-TiO2. Characteristic times for the various steps in the mechanism are given to the right of each step.
electron–holes separation
| S + hv → S1* (fs) | (1) |
| S1* → eCBI− + S+ (fs) | (2) |
electron trapping
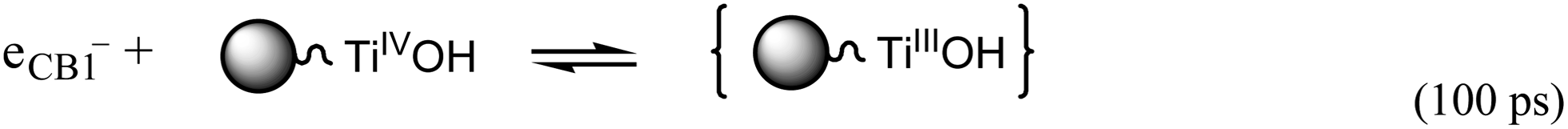 | (3) |
electron–holes recombination
 | (4) |
interfacial electron transfer
 | (5) |
| S+ + Red → S + Red˙+ (100 ns) | (6) |
 represents the primary hydrated surface functionality of TiO2, eCBI− is a conduction-band electron, Red is an electron donor (i.e., reductant),
represents the primary hydrated surface functionality of TiO2, eCBI− is a conduction-band electron, Red is an electron donor (i.e., reductant),  is the surface-trapped CB1 electron.
is the surface-trapped CB1 electron.
The pathways and time scales for relaxation of excited state of CuTCPP-TiO2 as discussed in the preceding paragraphs are represented schematically in Fig. 12. When the visible light irradiates on the surface of CuTCPP-TiO2 nanoparticles, the electrons in CuTCPP can be excited from the S0 level to the “S1” or “S2” of the porphyrin ring (π*). Then the electron in S2 state comes back to the original S0 level accompanied by the photon emission whereas the electron in S1 state have two pathways: leaves for the P25 CB1 via the Ti–O bond and intersystem crossing to T1. The dominant channel for Qx relaxation is intersystem crossing to T1, and the lifetime of the T1 state is reported to be milliseconds in degassed solvents.36 Subsequently, the electron in CB1 was trapped in 100 ps by the reaction as shown in eqn (3). Then the trapped electron  will, either recombine with holes depicted as eqn (4), or interfacial transfer to encounter with O2 adsorbed on the surface to reduce to O2˙−, which can further transform into H2O2 and ˙OH, resulting in the oxidation of MB finally. Meanwhile, the reactive holes S+ can oxidize MB to its radical cation either directly or through a primarily formed ˙OH produced by the oxidation of ubiquitous water.
will, either recombine with holes depicted as eqn (4), or interfacial transfer to encounter with O2 adsorbed on the surface to reduce to O2˙−, which can further transform into H2O2 and ˙OH, resulting in the oxidation of MB finally. Meanwhile, the reactive holes S+ can oxidize MB to its radical cation either directly or through a primarily formed ˙OH produced by the oxidation of ubiquitous water.
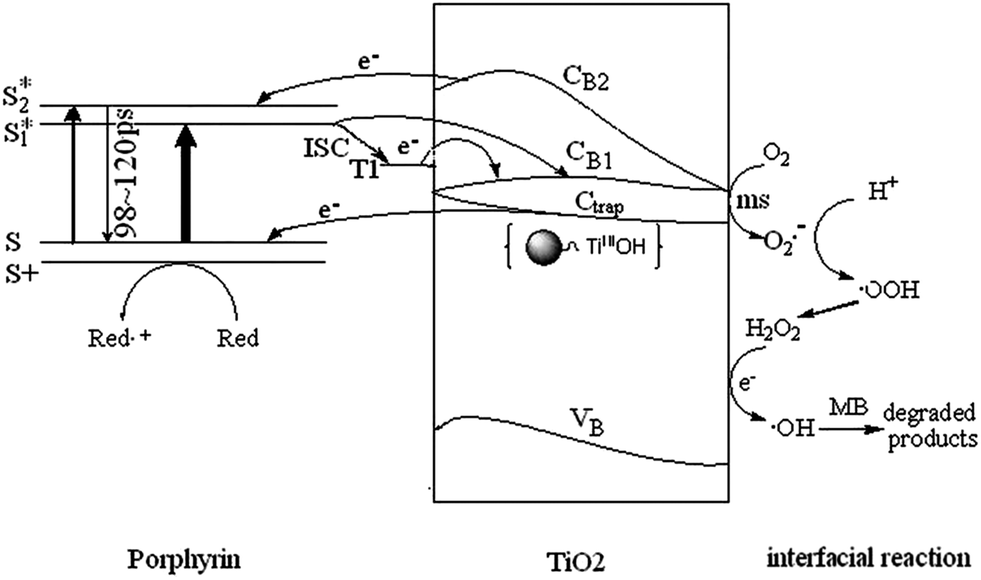 | ||
| Fig. 12 Schematic presentations of energy levels, basic photoinduced electron decay process, and photoinduced MB degradation mechanism. | ||
According to the mechanism illustrated in Fig. 12 and eqn (4) and (5), the overall quantum efficiency for interfacial charge transfer is determined by two critical processes. They are the competition between electron–holes recombination and trapping (picoseconds to nanoseconds) followed by the competition between trapped electron recombination and interfacial charge transfer (microseconds to milliseconds). The prolonged lifetime in trapped electron–holes recombination favors the interfacial charge transfer and enhances the degradation efficiency.
5. Conclusions
CuTCPP were chemically sensitized on the surface of TiO2 to act as visible light antenna and to modify the photoresponse properties of TiO2 particles. The TCSPC technique combined with their fluorescence spectra revealed that the S2–S0 fluorescence of CuTCPP is decreased in intensity by TiO2 and blue shifted by 18.7 nm in wavelength, with the lifetime prolonged. Ti 2p and O 1s XPS spectra of pure TiO2 and CuTCPP-TiO2 provide a direct demonstration for the formation of Ti–O–CO bond.
The CuTCPP-TiO2 exhibits better photoactivity under visible light irradiation than that of pure TiO2. The photocatalytic degradation kinetics of MB fitted well with Langmuir–Hinshelwood mode. This efficiency in the MB-photocatalytic activity of CuTCPP-TiO2 could be due to a combined effect of the potential photosensitivity of the CuTCPP-TiO2 band gap, the increase in MB adsorption capacity and the prolonged lifetime for electron transfer from photo-excited porphyrin to the surface of TiO2.
The development of the porphyrin-based photocatalyst provides an alternative approach in harnessing solar visible light. Further investigations about the adsorption and photocatalysis behaviors should be made to obtain deeper insight into the cooperative action mechanism between them.
Acknowledgements
This work was supported by grants from Alexander von Humboldt Foundation, National Natural Science Foundation of China (no. 21271155 and 21273202), Natural Science Foundation of Zhejiang Province (no. LY13B030009) and Zhejiang SCI-TECH University for 521 Distinguished Scholars scheme.References
- J. E. Johnston and J. M. Gibson, Environ. Sci. Technol., 2013, 47, 5595–5602 CrossRef CAS PubMed.
- X. M. Wang, L. J. Xing, T. L. Qiu and M. L. Han, Environ. Sci. Pollut. Res., 2013, 20, 2236–2243 CrossRef CAS PubMed.
- G. M. Ratnamala, K. V. Shetty and G. Srinikethan, Water, Air, Soil Pollut., 2012, 223, 6187–6199 CrossRef CAS.
- P. Punamiya, D. Sarkar, S. Rakshit and R. Datta, J. Environ. Qual., 2013, 42, 1449–1459 CrossRef CAS PubMed.
- O. F. Olorundare, R. W. M. Krause, J. O. Okonkwo and B. B. Mamba, Phys. Chem. Earth, 2012, 50–52, 104–110 CrossRef PubMed.
- R. M. Hlihor and M. Gavrilescu, Environ. Eng. Manage. J., 2009, 8, 353–372 CAS.
- U. C. Panda, P. Rath, S. Bramha and K. C. Sahu, J. Coastal Res., 2010, 26, 860–868 CrossRef CAS.
- L. Vezzaro, E. Eriksson, A. Ledin and P. S. Mikkelsen, Water Res., 2012, 46, 6891–6903 CrossRef CAS PubMed.
- S. Sarkar, A. Makhal, T. Bora, K. Lakhsman, A. Singha, J. Dutta and S. K. Pal, ACS Appl. Mater. Interfaces, 2012, 4, 7026–7034 Search PubMed.
- S. Ardo, D. Achey, A. J. Morris, M. Abrahamsson and G. J. Meyer, J. Am. Chem. Soc., 2011, 133, 16572–16580 CrossRef CAS PubMed.
- D. Li, W. J. Dong, S. M. Sun, Z. Shi and S. H. Feng, J. Phys. Chem. C, 2008, 112, 14878–14882 CAS.
- J. Ananpattarachai, P. Kajitvichyanukul and S. Seraphin, J. Hazard. Mater., 2009, 168, 253–261 CrossRef CAS PubMed.
- J. H. Cai, J. W. Huang, H. C. Yu and L. N. Ji, Int. J. Photoenergy, 2012, 348292 Search PubMed.
- M. K. Panda, K. Ladomenou and A. G. Coutsolelos, Coord. Chem. Rev., 2012, 256, 2601–2627 CrossRef CAS PubMed.
- M. Pelaez, N. T. Nolan, S. C. Pillai, M. K. Seery, P. Falaras, A. G. Kontos, P. S. M. Dunlop, J. W. J. Hamilton, J. A. Byrne, K. O'Shea, M. H. Entezari and D. D. Dionysiou, Appl. Catal., B, 2012, 125, 331–349 CrossRef CAS PubMed.
- C. Wang, X. X. Ma, J. Li, L. Xu and F. X. Zhang, J. Mol. Catal. A: Chem., 2012, 363, 108–114 CrossRef PubMed.
- M. J. Griffith, K. Sunahara, P. Wagner, K. Wagner, G. G. Wallace, D. L. Officer, A. Furube, R. Katoh, S. Mori and A. J. Mozer, Chem. Commun., 2012, 48, 4145–4162 RSC.
- W. J. Sun, J. Li, G. P. Yao, M. Jiang and F. X. Zhang, Catal. Commun., 2011, 16, 90–93 CrossRef CAS PubMed.
- W. Kim, J. Park, H. J. Jo, H. J. Kim and W. Choi, J. Phys. Chem. C, 2008, 112, 491–499 CAS.
- G. Mele, R. Del Sole, G. Vasapollo, G. Marci, E. Garcia-Lopez, L. Palmisano, J. M. Coronado, M. D. Hernandez-Alonso, C. Malitesta and M. R. Guascito, J. Phys. Chem. B, 2005, 109, 12347–12352 CrossRef CAS PubMed.
- A. Marcelli, I. J. Badovinac, N. Orlic, P. R. Salvi and C. Gellini, Photochem. Photobiol. Sci., 2013, 12, 348–355 CAS.
- A. Marcelli, P. Foggi, L. Moroni, C. Gellini and P. R. Salvi, J. Phys. Chem. A, 2008, 112, 1864–1872 CrossRef CAS PubMed.
- V. N. Knyukshto, E. I. Sagun, A. M. Shulga, S. M. Bachilo and E. I. Zenkevich, Chem. Phys. Rep., 1999, 18, 855–872 Search PubMed.
- M. R. Hoffmann, S. T. Martin, W. Y. Choi and D. W. Bahnemann, Chem. Rev., 1995, 95, 69–96 CrossRef CAS.
- J. R. McCarthy and R. Weissleder, ChemMedChem, 2007, 2, 360–365 CrossRef CAS PubMed.
- K. Rurack, Spectrochim. Acta, Part A, 2001, 57, 2161–2195 CrossRef CAS.
- D. F. Marsh and L. M. Mink, J. Chem. Educ., 1996, 73, 1188–1190 CrossRef CAS.
- J. M. Wan, H. G. Wang, Z. Z. Wu, Y. C. Shun, X. M. Zheng and D. L. Phillips, Phys. Chem. Chem. Phys., 2011, 13, 10183–10190 RSC.
- H. G. Wang, J. Xu, J. M. Wan, Y. Y. Zhao and X. M. Zheng, J. Phys. Chem. B, 2010, 114, 3623–3632 CrossRef CAS PubMed.
- L. H. Yu, J. Y. Xi, M. D. Li, H. T. Chan, T. Su, D. L. Phillips and W. K. Chan, Phys. Chem. Chem. Phys., 2012, 14, 3589–3595 RSC.
- A. Kathiravan, P. S. Kumar, R. Renganathan and S. Anandan, Colloids Surf., A, 2009, 333, 175–181 CrossRef CAS PubMed.
- T. Yoshihara, R. Katoh, A. Furube, Y. Tamaki, M. Murai, K. Hara, S. Murata, H. Arakawa and M. Tachiya, J. Phys. Chem. B, 2004, 108, 3817–3823 CrossRef CAS.
- H. Z. Yu, J. S. Baskin and A. H. Zewail, J. Phys. Chem. A, 2002, 106, 9845–9854 CrossRef CAS.
- A. Houas, H. Lachheb, M. Ksibi, E. Elaloui, C. Guillard and J. M. Herrmann, Appl. Catal., B, 2001, 31, 145–157 CrossRef CAS.
- G. Spoto, C. Morterra, L. Marchese, L. Orio and A. Zecchina, Vacuum, 1990, 41, 37–39 CrossRef CAS.
- L. Pekkarinen and H. Linschitz, J. Am. Chem. Soc., 1960, 82, 2407–2411 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2014 |